
2 Chapter 2. DNA Structure and Replication
Albert B. Flavier
Chapter Outline
2.2. Chemical structure of DNA
2.5. Regulation of Chromosome Replication
2.6. Antibiotics that inhibit DNA replication
Learning Objectives
- Be able to name scientists and their contributions to our knowledge of DNA as genetic material.
- Be able to describe the chemical composition and structure of DNA
- Be able to draw and describe the process of DNA replication, the proteins required, and their roles during replication.
- Describe some important uses of DNA polymerases in biotechnology.
Central Dogma of Molecular Biology
In all domains of life, the central dogma of molecular biology is a theory that describes how the genetic information in DNA (or genotype) is transcribed to mRNA, and the information in mRNA translated to chains of amino acids in proteins (Fig. 2.1). In the end, these proteins and the influence of the cellular and organismal environment determine the phenotype or physical traits of the cell or organism. DNA replication is required for the passage of information from parental to daughter cells. The dogma applies from the simplest bacteria to the most complex eukaryotes. However, we know now that RNA can also be reverse transcribed to DNA. There are also non-coding RNAs which could affect gene expression and phenotypes. Because of their less complex organization, much of what we know about DNA and its replication, transcription, translation and other molecular biology processes started with studies done in bacteria (and their viruses).
Aside from their use as mini factories for useful industrial products, the bacterium, Escherichia coli, and the yeast, Saccharomyces cerevisiae, because of their simple cellular and biochemical organization, and fast growth, also serve as model organisms for understanding biological processes in higher orders of life. The single chromosome in E. coli, small size of the genome (4600 base pairs), and minimal duplication of genes and gene functions, made facile the dissection of the effects of certain mutations in cellular processes. Much of our current understanding of genetics, cellular organization, protein functions, and biochemical pathways are derived, therefore, from studies involving E. coli and S. cerevisiae.
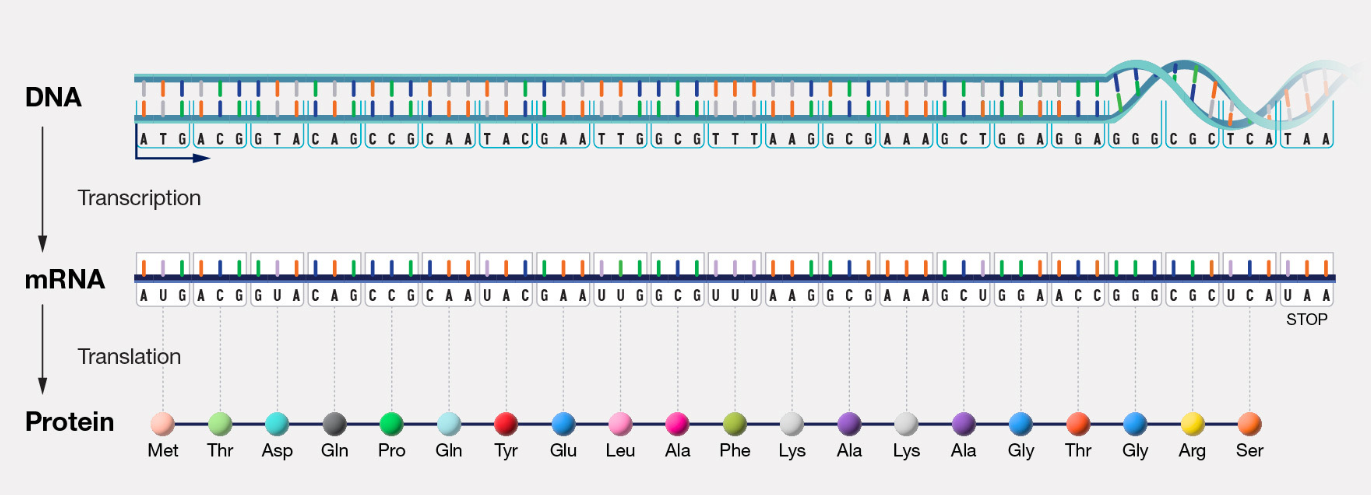
Figure 2.1. Central Dogma of Molecular Biology. Information in DNA is used to transcribe mRNA which is translated into protein. CC: NIH-National Human Genome Research Institute (public domain).
Watch:
2.1 DNA as unit of heredity
The Austrian monk, Gregor Mendel, experimenting with garden peas in the 1860s, postulated the existence of a determinants responsible for the inheritance of traits from one generation and to successive generations. These determinants of inheritance were later referred to as genes, the unit of heredity. It took many more decades and researchers before the chemical nature of genes will be known.
Table 2.1 Timeline and important contributions to DNA structure and DNA as genetic material.
|
Year |
Scientist |
Discovery |
|
1865 |
Mendel |
Father of genetics. Traits are passed down from parents to offsprings |
|
1869 |
Miescher |
Isolated DNA (nuclein) from nucleus of white blood cells, then other cells |
|
1928 |
Griffith |
A substance in killed smooth S. pneumoniae is able to transform rough S. pneumoniae to smooth phenotype |
|
1929 |
Levene |
DNA nucleotides are made up of nitrogenous bases, deoxyribose, and phosphates |
|
1944 |
Avery, MacCleod, McCarty |
The S. pneumoniae transforming principle is DNA and is destroyed by DNAse |
|
1950 |
Chargaff |
Amount of adenine is equal to thymine, and cytosine equal to guanine in DNA |
|
1952 |
Hershey and Chase |
DNA is genetic material that contains instructions for bacteriophage replication inside E. coli |
|
1953 |
Franklin and Wilkins |
X-ray crystallography suggested helical nature of DNA |
|
1953 |
Watson and Crick |
Proposed the double stranded, helical structure of DNA |
|
1958 |
Meselson and Stahl |
DNA replication is semi-conservative |
In 1869, while attempting to understand the molecular basis of life of cells, Friedrich Miescher isolated a phosphorus-rich compound, distinct from proteins, from the nuclei of white blood cells in pus from surgical bandages. He referred to the compound as nuclein. Later, he showed that nuclein can be isolated from all types of cells and abundantly in salmon sperm which has a large nuclei and other vertebrate sperm. Because the compound was acidic, it was referred to later as nucleic acid. While Miescher did the pioneering work on the isolation of what will be called later as DNA, demonstration of the role of DNA in the inheritance of organismal traits was to come much later. Proteins, because of their more complex composition compared to nucleic acids, were initially thought to be the inherited factors responsible for passing many different traits to subsequent generations.
In the early part of the twentieth century, before the advent of antibiotics, pneumococcal infections caused by the Gram-positive bacterium, Streptococcus pneumoniae, claimed many lives. As a result, researchers on both sides of the Atlantic were actively engaged in studying the bacterium. S. pneumoniae is common in nose and throat of up to 90% of the population, but it can get into the lungs where it can cause fevers and respiratory complications that can lead to deaths. Some strains can also infect the spinal cord and brain where they can cause pneumococcal meningitis characterized by headache and confusion.
Early scientists have observed two strains of S. pneumoniae on agar plates, where one strain forms smooth, convex, and shiny colonies (S-strain), while the second strain forms rough and flat looking colonies (R-strain) (See Figure 3 Belanger et al. ). The smooth appearance is due to the production of a capsule consisting of exopolysaccharides. The capsule is a major virulence factor which allows attachment of S. pneumoniae to host cells and protection from immune system attack.
In 1928, Frederick Griffith conducted experiments that showed the smooth variant is virulent and kills mice when injected, while the rough variant is not virulent (Fig. 2.2). He showed that heating the S strain at 60oC led to loss of virulence and no S strains were recoverable from mice injected with the heat-killed cells. Griffith found that co-injecting heat-treated S variants with the non-virulent R variants killed mice. From the killed mice, he was able to isolate live, S variants. In contrast, the R variant by itself does not revert to the S-type. This led him to propose that there exists a substance from the killed S-cells that are able to transform R cells into S cells. Griffith speculated then that the transforming factors are the polysaccharides in the capsule of the S cells. Scientists were convinced that proteins are the more likely determinants (carriers) of traits (genetic information) because there are many more combinations of 20 different amino acids that can be present in proteins, than combinations that can be generated from only four different nucleotides.
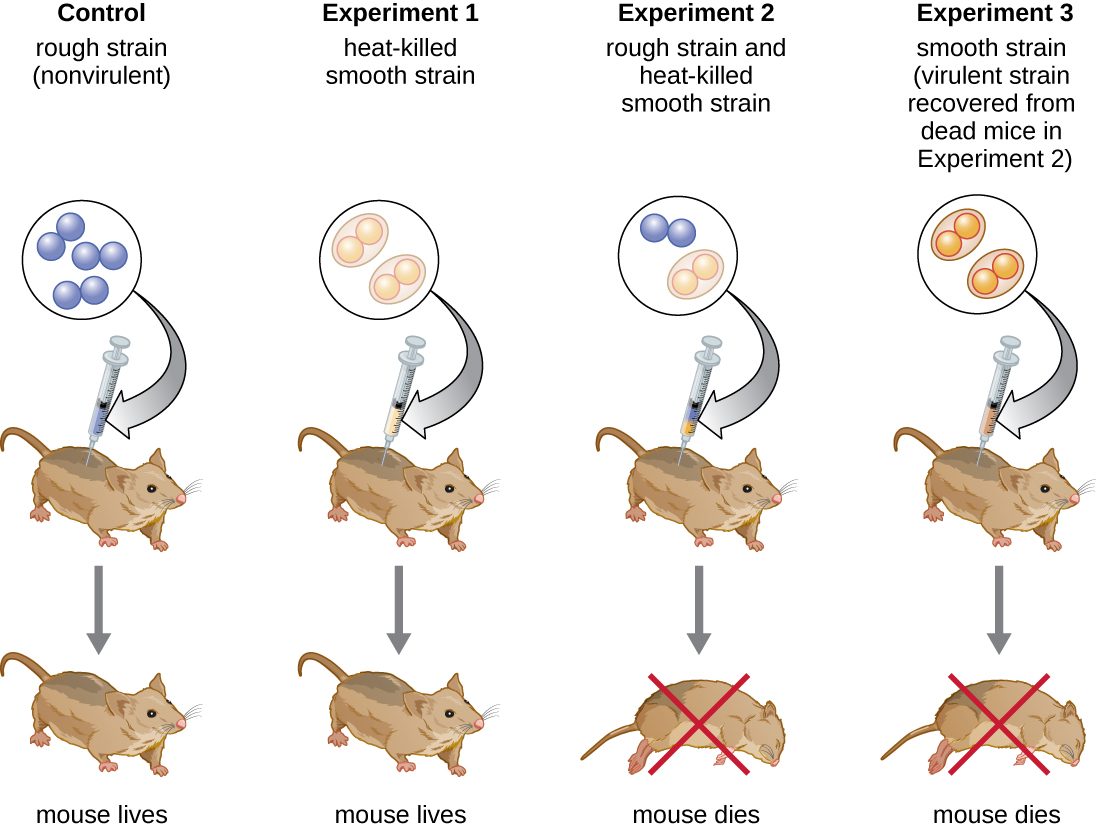
Figure 2.2. In his famous series of experiments, Griffith used two strains of S. pneumoniae. The S strain is pathogenic and causes death when injected in mice (not shown). Mice injected with the nonpathogenic R strain (control) or the heat-killed S strain survive (Experiment 1). However, a combination of the heat-killed S strain and the live R strain causes many mice to die (Experiment 2). S strain was recovered from the dead mouse (Experiment 3) suggesting that something had passed from the heat-killed S strain to the R strain, transforming the R strain into an S strain in the process. CC BY-SA 4.0 from Parker.
Scientists were convinced that proteins are the more likely determinants (carriers) of traits (genetic information) because there are many more combinations of 20 different amino acids that can be present in proteins, than combinations that can be generated from only four different nucleotides.
Oswald Avery, Colin MacLeod, and Maclyn McCarty took Griffith’s experiments a step further by looking for the chemical from heat-killed S cells that transform R to S cells. They prepared extracts from the heat-killed S cells (Fig 2.3) and showed that treatment of the extracts with deoxyribonuclease (DNase), an enzyme that degrades DNA, led to loss of the ability of the extract to transform R cells, while degrading proteins with proteinases, and RNA with ribonucleases (RNase), still enabled transformation. Use of heat inactivated DNAse did not lead to a loss of the transforming principle. In another experiment, they purified DNA from the S-cell extracts and showed that DNA alone is able to transform the R to S cells. These experiments provided proof that DNA is the transforming principle, not RNA nor protein.
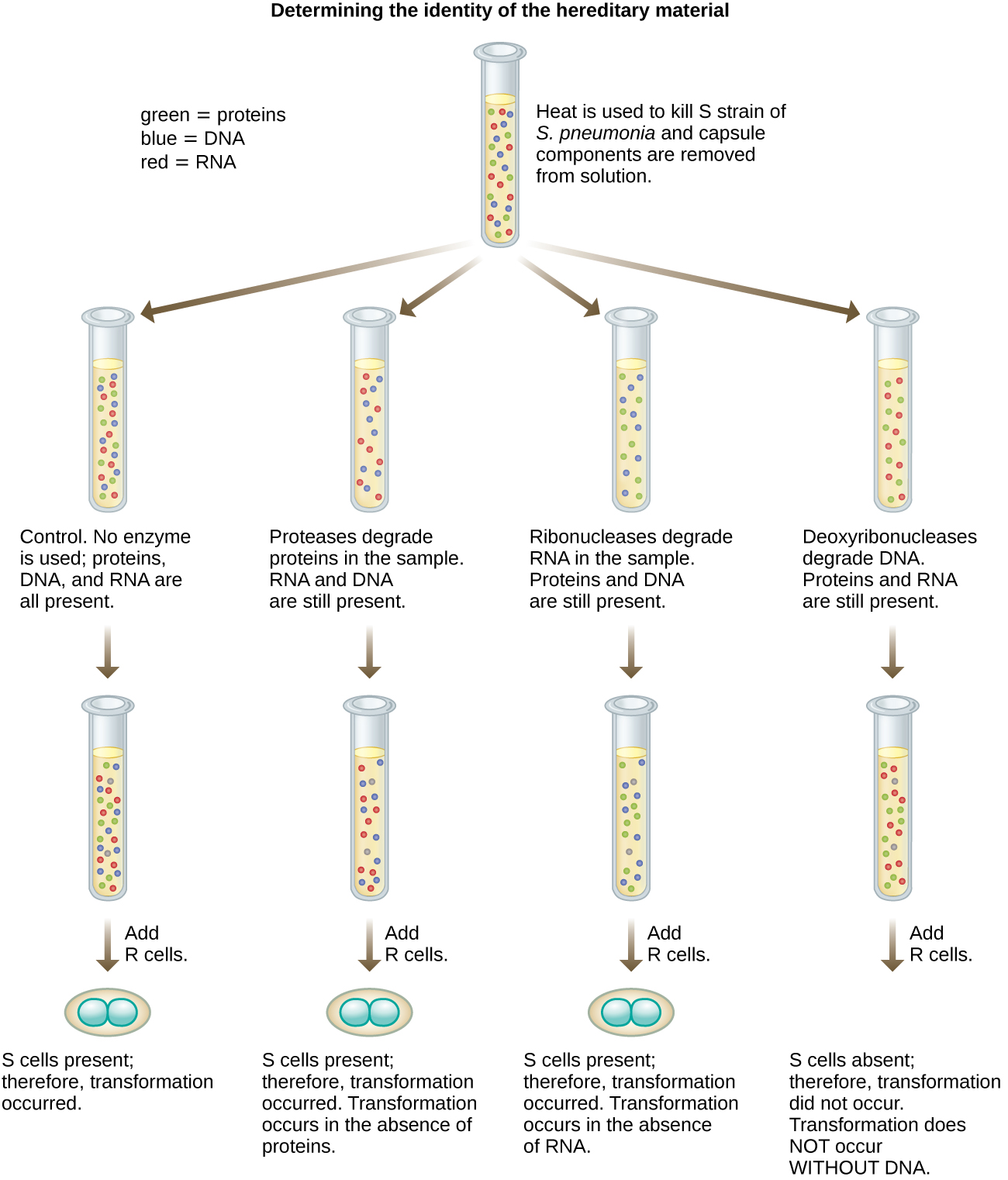
Figure 2.3. Avery, MacLeod, and McCarty’s experiment that showed that treatment with deoxyribonuclease destroyed the transforming principle. CC BY-SA 4.0 from Parker.

Figure 2.4 (a) Electron micrograph of a bacteriophage (a virus that infects bacteria) infecting a bacterial cell. (b) Illustration of a bacteriophage in the micrograph. (Credit a: modification of work by U.S. Department of Energy, Office of Science, LBL, PBD. CC-SA-4.0 Parker et al. 2016.
Further proof that the genetic material is indeed DNA came from studies using bacteriophages (Fig. 2.4). Bacteriophages are viruses that are able to infect and replicate inside bacterial cells that then lyze the infected cells to release thousands of phage progenies. To be able to replicate, the genetic material has to be injected inside the bacteria. T2 bacteriophages only have a protein capsid and DNA inside the capsid, i.e., they have no carbohydrates or RNA. In 1952, Alfred Hershey and Martha Chase sought to find out if it’s the protein or DNA component of the T2 bacteriophage which is injected in E. coli and responsible for replication (Fig. 2.5). E. coli was grown for 4 hours in growth media containing radioisotopes of either sulfur (35S) or phosphorus (32P), then infected with T2. Upon replication inside E. coli, the 35S would get incorporated only in the protein coat of T2 since the amino acids cysteine and methionine have sulfur. The 32P, in a separate culture media, will only get incorporated in DNA, which has phosphate in its backbone, while there’s no phosphorus in amino acids. The 35S and 32P-labelled T2 were then allowed to infect cultures of E. coli grown separately in fresh, non-radioactive media, then the empty phages detached by agitating the phage-bacteria suspension in a blender. The suspensions were then centrifuged where the bacteria ended up in the pellet while the smaller and lighter phages remained in the supernatant. 32P was found in the pelleted E. coli, while 35S remained mostly in the supernatant (with the phages), evidence that DNA is the genetic material that is injected into and needed for T2 replication inside E. coli, but proteins most likely are not.
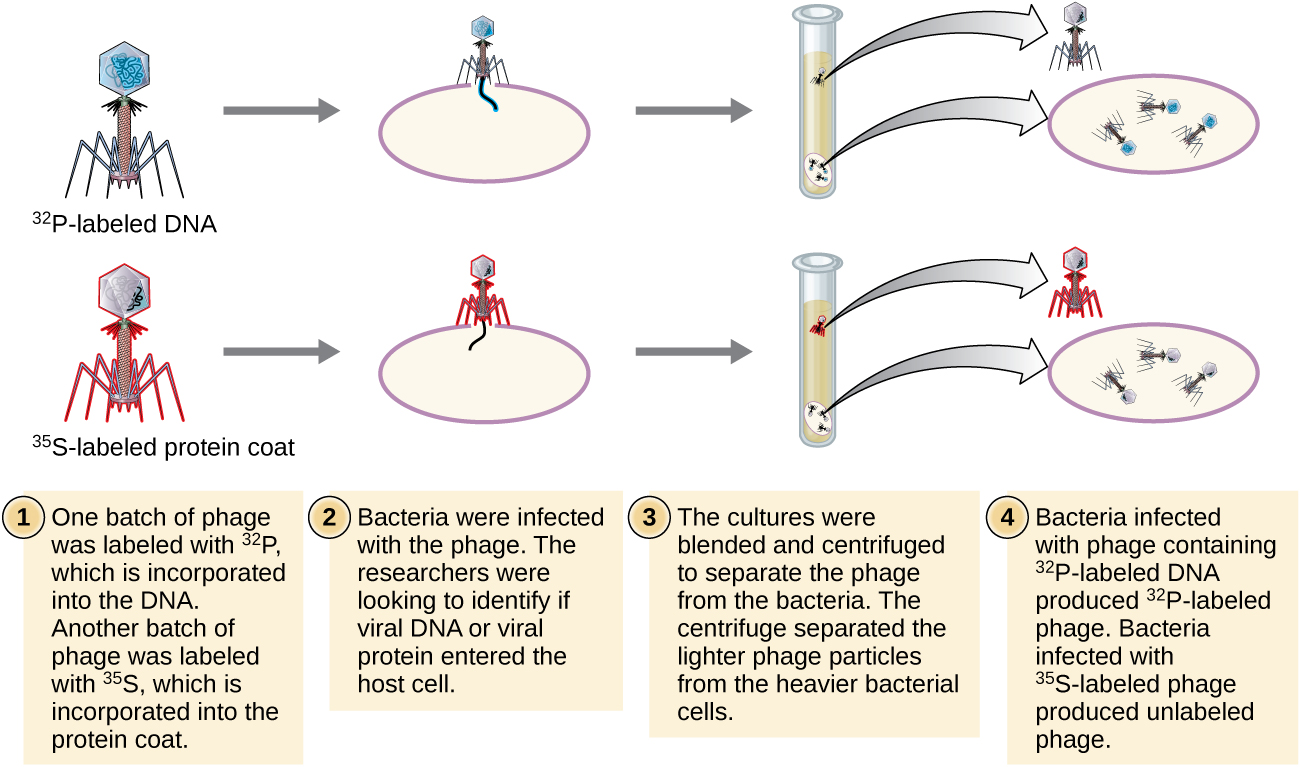
Figure 2.5. Martha Chase and Alfred Hershey conducted an experiment separately radiolabeling T2 bacteriophage DNA with 32P and proteins with 35S and showed 32P labelled DNA got inside E. coli while 35S labelled proteins remained outside of E. coli. CC BY-SA 4.0 from Parker.
2.2 Chemical Structure of DNA
In 1929, the Lithuania-born biochemist, Phoebus Levene, showed that DNA consists of a string of nucleotides, where each nucleotide consists of one of four different nitrogenous bases (adenine, guanine, cytosine and thymine), a phosphate group, and the unique sugar, deoxyribose (Fig. 2.6). In 1950, Erwin Chargaff, described that there are differences in nucleotide composition and arrangement in the DNA of different organisms. However, despite the differences in overall composition, the amount of adenine (A) is always equal to that of thymine (T), while the amount of guanine (G) is always equal to that of cytosine (C). He was not able to give much significance to the pairing of A with T and G with C.
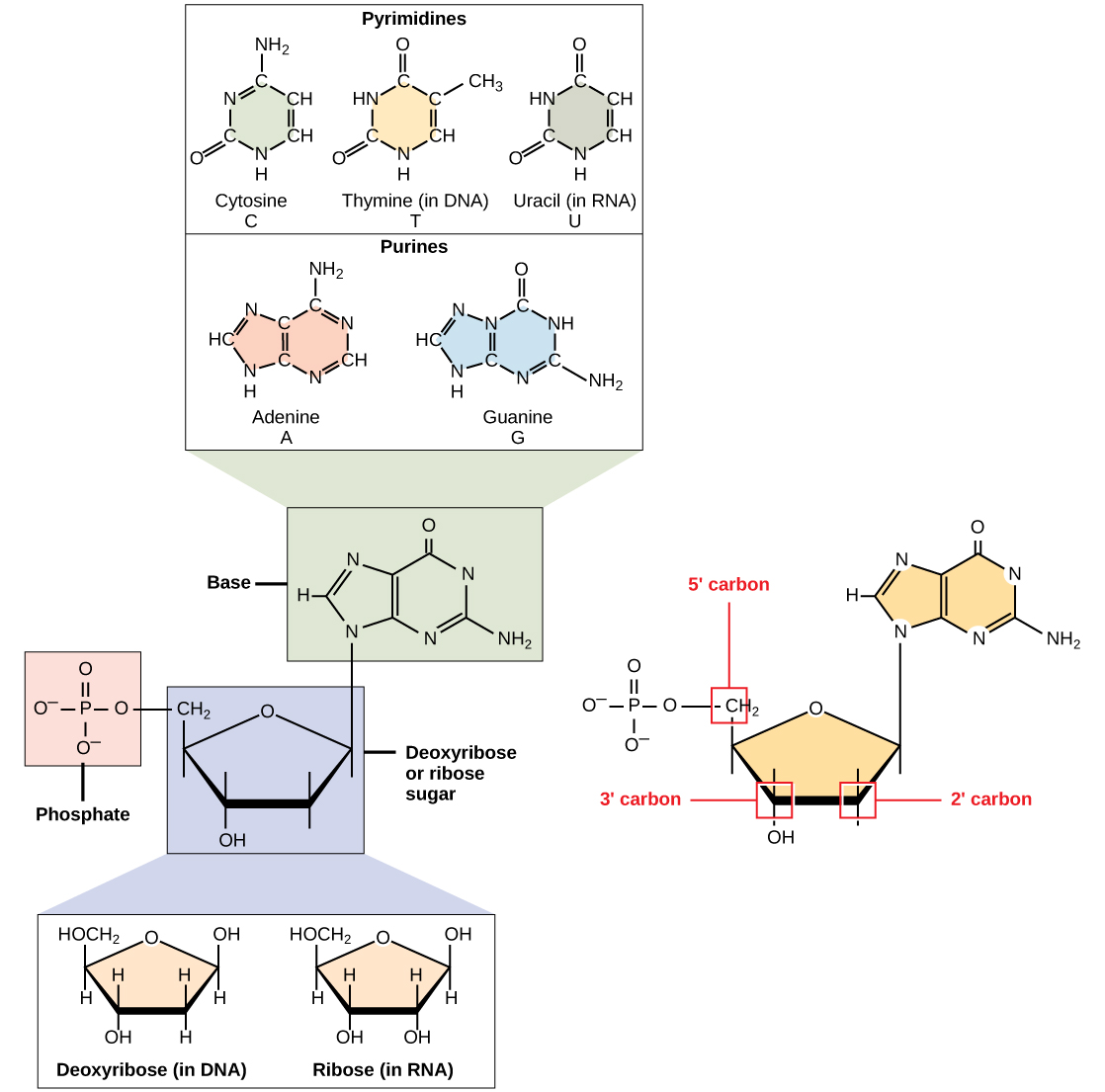
Figure 2.6. Five different nucleotides found in DNA or RNA showing the three different components of each nucleotide – the pentose sugar which can be deoxyribose (in DNA) or ribose (in RNA), the phosphate group attached to the 5’ carbon, and the nitrogenous bases. CC0 1.0 from OpenStax

Figure 2.7. Three scientists whose works contributed to the double-helical model of DNA. Attribution: James Watson, Francis Crick, and Rosalind Franklin.
In 1953, a year after Hershey and Chase’ experiments, the double helical nature of DNA and its role in cellular replication was proposed by James Watson and Francis Crick (Fig. 2.7). Later, Rosalind Franklin and Maurice Wilkins, examined the 3-dimensional crystal structure of DNA using X-ray diffraction. Finally, in 1953, based on Chargaff’s rule and Franklin and Wilkins observations, Watson and Crick proposed the double helical model of DNA. In the Watson and Crick model, the two intertwined polynucleotides are joined by hydrogen bonds – two H-bonds between A and T, and three H-bonds between G and C (Fig. 2.8), which is in agreement with Chargaff’s base-pairing rule. The successive nucleotides in each strand are linked by the 5’ phosphate and 3’ hydroxy groups of the deoxyribose. The two polynucleotide strands are anti-parallel where the 5’ end of one strand is matched with the 3’ end of the second strand and vice-versa. The complementarity of the bases in opposite strands of DNA in the Watson and Crick model provided a mechanism by which one strand contains information which can serve as the template for the synthesis of a new, second strand during replication, and provides an explanation for how genes, and therefore, traits can be inherited by daughter cells after cell division.
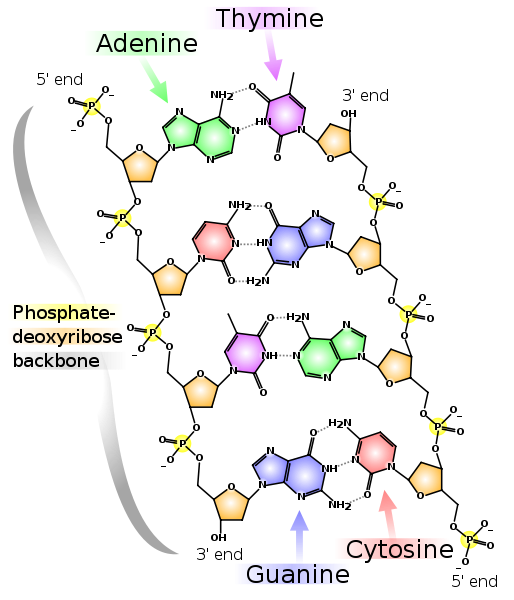
Figure 2.8. Double-stranded nature of DNA as proposed by Watson and Crick. The nucleotides in the polymer are linked by phosphate-deoxyribose backbone. Each strand exhibits polarity with a phosphate group in the 5’ carbon and OH group in the 3’ carbon. The two strands are anti-parallel with one strand oriented in 5’ to 3’ direction and the complementary strand oriented in 3’ to 5’ direction. The bases in the two opposite strands are linked by hydrogen bonds. CC-0 by Madeleine Price Ball
2.3 The Bacterial Genome
A genome is the sum total of an organism’s genetic information which includes both coding and non-coding DNA. Most bacteria and archaea have single, circular chromosome of supercoiled DNA (Fig. 2.9). There are, however, species with multiple chromosomes, as well as those with linear DNA or both. Bacteria lack nuclei so the bacterial chromosome(s) is/are found floating freely in the cytoplasm.
In bacteria and some yeasts, the genome often includes smaller pieces of circular DNA called plasmids (Fig. 2.9). Plasmids replicate autonomously from the chromosome, though often synchronized with the chromosome. Plasmids typically contain stress or survival related genes (survival in presence of toxic chemicals, pH, high salt, mercury). They also carry genes involved in antibiotic synthesis to kill off competing microbes, and antibiotic resistance genes for resistance to the same antibiotic which they produce, or to tolerate antibiotics which are produce by plants, animals, or other microbes that are trying to eliminate them.
Plasmids can be readily transferred to other non-daughter cells in a population by horizontal gene transfer and unlike the chromosome, plasmids can exist in high copy numbers within the cell versus only 1 or 2 for the chromosome. In recombinant protein production. More mRNA > more protein. Plasmids will be discussed in more detail in Chapter 8.
|
|
|
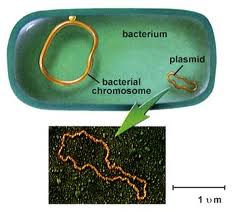
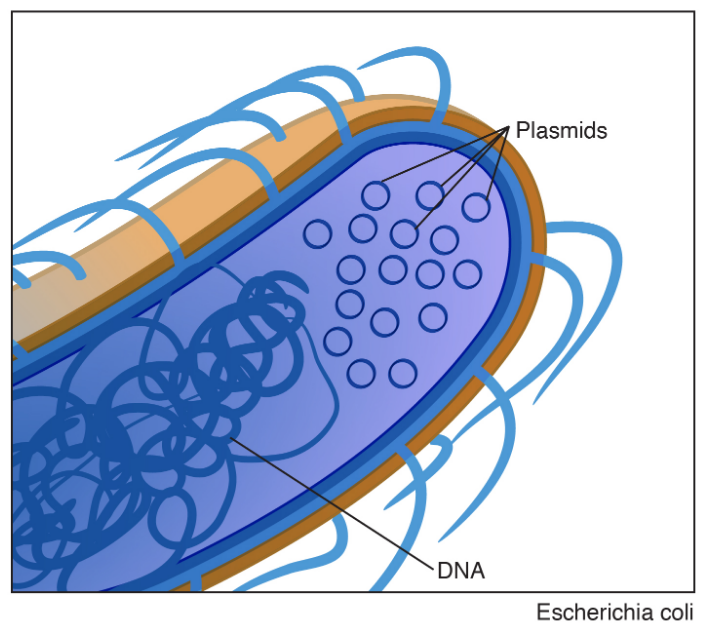
Figure 2.9. Bacteria genome consisting of a single circular chromosome and a smaller plasmid. Left image taken from sandwalk/blog. Right image is public domain.
The genome contains many genes which code for proteins, or RNA, that are used to build and maintain the various processes occurring within and between cells including their own propagation from parent to offspring. A gene contains a portion that is transcribed to RNA and untranscribed regions 5’ and 3’ of the RNA coding region (Fig. 2.10). These untranscribed regions are involved in regulating and terminating transcription. The RNA that is transcribed can code for a protein. There are also untranslated sequences 5’ and 3’ of the protein coding region and these regions are involved in regulating and terminating translation of the RNA.
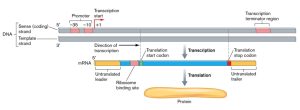
Figure 2.10. Prokaryotic genes are relatively simple and composed of: 1) a 5′ promoter/binding region for the initiation of transcription, 2) a long unbroken region that is used as a template for RNA synthesis and which contains the protein coding region/s (ORF), and 3) a trailing 3′ region of non-coding DNA that adds stability to the nascent RNA molecule. CC SA-3.0 Unported by Slonczewski and Foster
2.4 DNA Replication
The elucidation of the structure of the double helix by James Watson and Francis Crick in 1953 provided a hint as to how DNA is copied during the process of replication. Separating the strands of the double helix would provide two templates for the synthesis of new complementary strands, but exactly how new DNA molecules were constructed was still unclear. In one model, semiconservative replication, the two strands of the double helix separate during DNA replication, and each strand serves as a template from which the new complementary strand is copied; after replication, each double-stranded DNA includes one parental or “old” strand and one “new” strand. There were two competing models also suggested: conservative and dispersive (Fig. 2.11).
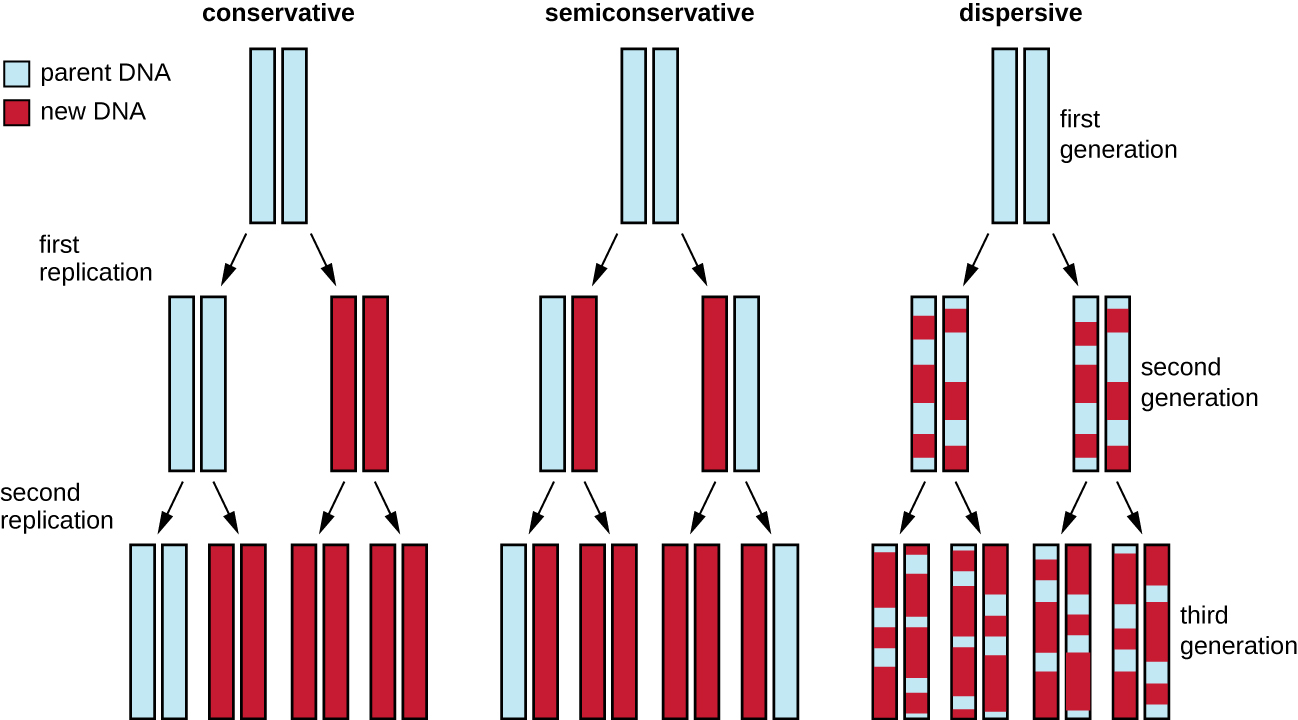
Figure 2.11 There were three models suggested for DNA replication. In the conservative model, parental DNA strands (blue) remained associated in one DNA molecule while new daughter strands (red) remained associated in newly formed DNA molecules. In the semiconservative model, parental strands separated and directed the synthesis of a daughter strand, with each resulting DNA molecule being a hybrid of a parental strand and a daughter strand. In the dispersive model, all resulting DNA strands have regions of double-stranded parental DNA and regions of double-stranded daughter DNA. CC-SA-4.0 Parker et al 2016
Matthew Meselson (1930–) and Franklin Stahl (1929–) devised an experiment in 1958 to test which of these models correctly represents DNA replication (Fig. 2.12). They grew E. coli for several generations in a medium containing a “heavy” isotope of nitrogen (15N) that was incorporated into nitrogenous bases and, eventually, into the DNA. This labeled the parental DNA. The E. coli culture was then shifted into a medium containing 14N and allowed to grow for one generation. The cells were harvested and the DNA was isolated. The DNA was separated by ultracentrifugation, during which the DNA formed bands according to its density. DNA grown in 15N would be expected to form a band at a higher density position than that grown in 14N. Meselson and Stahl noted that after one generation of growth in 14N, the single band observed was intermediate in position in between DNA of cells grown exclusively in 15N or 14N. This suggested either a semiconservative or dispersive mode of replication. Some cells were allowed to grow for one more generation in 14N and spun again. The DNA harvested from cells grown for two generations in 14N formed two bands: one DNA band was at the intermediate position between 15N and 14N, and the other corresponded to the band of 14N DNA. These results could only be explained if DNA replicates in a semiconservative manner. If DNA replication was dispersive, a single purple band positioned closer to the red 1414 would have been observed, as more 14 was added in a dispersive manner to replace 15. Therefore, the other two models were ruled out. As a result of this experiment, we now know that during DNA replication, each of the two strands that make up the double helix serves as a template from which new strands are copied. The new strand will be complementary to the parental or “old” strand. The resulting DNA molecules have the same sequence and are divided equally into the two daughter cells.
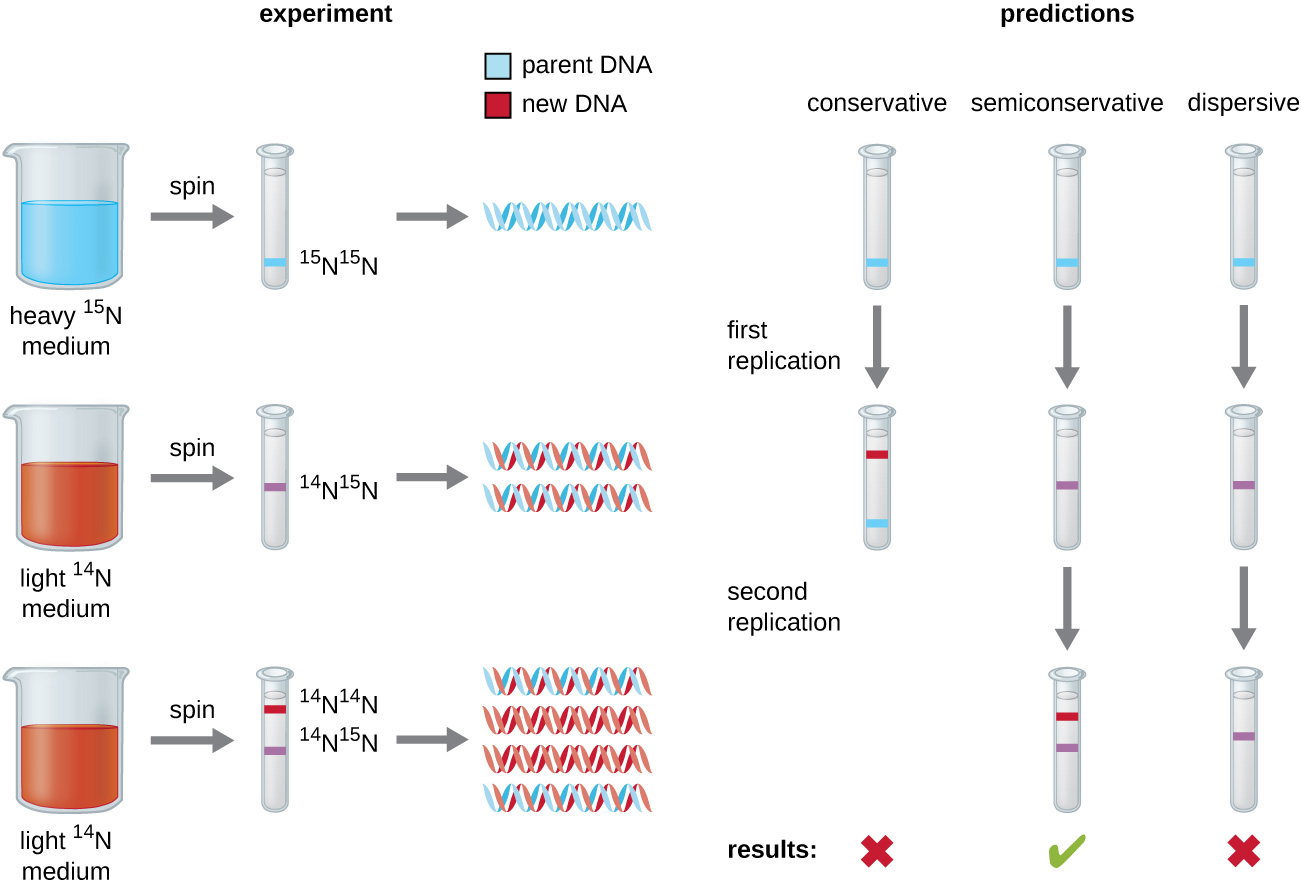
Figure 2.12. Meselson and Stahl experimented with E. coli grown first in heavy nitrogen (15N) then in 14N. DNA grown in 15N (blue band) was heavier than DNA grown in 14N (red band), and sedimented to a lower level on ultracentrifugation. After one round of replication, the DNA sedimented halfway between the 15N and 14N levels (purple band), ruling out the conservative model of replication. After a second round of replication, the dispersive model of replication was ruled out. These data supported the semiconservative replication model. CC-SA-4.0 Parker
DNA replication has been well studied in bacteria primarily because of the small size of the genome and the mutants that are available. E. coli has 4.6 million base pairs (Mbp) in a single circular chromosome and all of it is replicated in approximately 42 minutes, starting from a single origin of replication and proceeding around the circle bidirectionally (i.e., in both directions). This means that approximately 1000 nucleotides are added per second. The process is quite rapid and occurs with few errors.
DNA replication uses a large number of proteins and enzymes (Table 2.2). One of the key players is the enzyme DNA polymerase, also known as DNA pol. In bacteria, three main types of DNA polymerases are known: DNA pol I, DNA pol II, and DNA pol III. It is now known that DNA pol III is the enzyme required for DNA synthesis; DNA pol I and DNA pol II are primarily required for repair. DNA pol III adds deoxyribonucleotides each complementary to a nucleotide on the template strand, one by one to the 3’-OH group of the growing DNA chain. The addition of these nucleotides requires energy. This energy is present in the bonds of three phosphate groups attached to each nucleotide (a triphosphate nucleotide), similar to how energy is stored in the phosphate bonds of adenosine triphosphate (ATP) (Fig. 2.13). When the bond between the phosphates is broken and diphosphate is released, the energy released allows for the formation of a covalent phosphodiester bond by dehydration synthesis between the incoming nucleotide and the free 3’-OH group on the growing DNA strand.
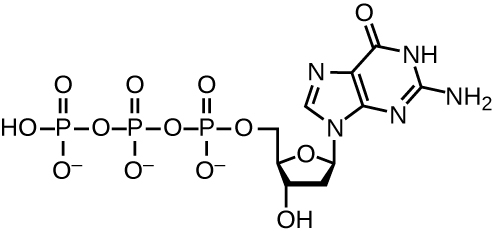 Figure 2.14. This structure shows the guanosine triphosphate deoxyribonucleotide that is incorporated into a growing DNA strand by cleaving the two end phosphate groups from the molecule and transferring the energy to the sugar–phosphate bond. The other three nucleotides form analogous structures. CC-SA-4.0 Parker
Figure 2.14. This structure shows the guanosine triphosphate deoxyribonucleotide that is incorporated into a growing DNA strand by cleaving the two end phosphate groups from the molecule and transferring the energy to the sugar–phosphate bond. The other three nucleotides form analogous structures. CC-SA-4.0 Parker
Initiation
The initiation of replication occurs at specific nucleotide sequence called the origin of replication, where various proteins bind to begin the replication process. E. coli has a single origin of replication (as do most prokaryotes), called oriC, on its one chromosome. The origin of replication is approximately 245 base pairs long and is rich in adenine-thymine (AT) sequences.
Chromosomal DNA is typically wrapped around histones (in eukaryotes and archaea) or histone-like proteins (in bacteria), and is supercoiled, or extensively wrapped and twisted on itself. This packaging makes the information in the DNA molecule inaccessible. For bacterial DNA replication to begin, the supercoiled chromosome is relaxed by topoisomerase II, also called DNA gyrase. An initiator protein, DnaA, in the presence of ATP as energy source, binds to repeat regions in the oriC, called DnaA boxes (Fig. 2.14), and with several accessory proteins, open up the double stranded origin by breaking the hydrogen bonds between the nitrogenous base pairs. Recall that AT sequences have fewer hydrogen bonds and, hence, have weaker interactions than guanine-cytosine (GC) sequences. The initiator complex recruits the hexameric enzyme helicase which further separates the DNA strands on opposite sides of the oriC. Loading of helicase also requires ATP hydrolysis. Thus, initiation of replication is energy demanding. As the DNA opens up, Y-shaped structures called replication forks are formed. Two replication forks are formed at the origin of replication, allowing for bidirectional replication and formation of a structure that looks like a bubble when viewed with a transmission electron microscope; as a result, this structure is called a replication bubble. The DNA near each replication fork is coated with single-stranded binding proteins to prevent the single-stranded DNA from reannealing back into a double helix.
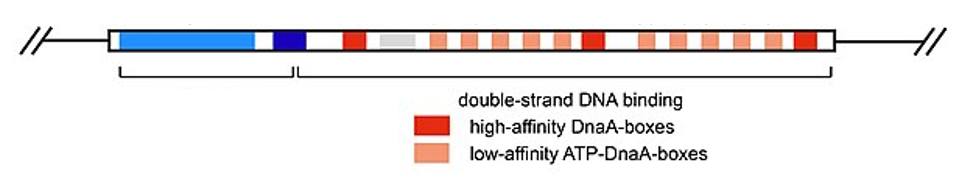
Figure 2.14. Structure of the E. coli oriC highlighting the DnaA boxes. CC SA 4.0 by Franziskableichert
Once single-stranded DNA is accessible at the origin of replication, DNA replication can begin. However, DNA polymerase III is able to add nucleotides only in the 5’ to 3’ direction (a new DNA strand can be only extended in this direction). This is because DNA polymerase requires a free 3’-OH group to which it can add nucleotides by forming a covalent phosphodiester bond between the 3’-OH end and the 5’ phosphate of the next nucleotide. This also means that it cannot add nucleotides if a free 3’-OH group is not available. The problem is solved with the help of a short RNA sequence that provides the free 3’-OH end. Because this sequence allows the start of DNA synthesis, it is appropriately called the primer. The primer is five to 10 nucleotides long and complementary to the parental or template DNA. It is synthesized by RNA primase, a special type of RNA polymerase. Unlike DNA polymerases, RNA polymerases do not need a free 3’-OH group to synthesize an RNA molecule. Now that the primer provides the free 3’-OH group, DNA polymerase III can now extend this RNA primer, adding DNA nucleotides one by one that are complementary to the parental strand.
Elongation
During elongation in DNA replication, the addition of nucleotides occurs at its maximal rate of about 1000 nucleotides per second. DNA polymerase III can only extend in the 5’ to 3’ direction, which poses a problem at the replication fork. The DNA double helix is antiparallel; that is, one strand is oriented in the 5’ to 3’ direction and the other is oriented in the 3’ to 5’ direction. During replication, one strand, which is complementary to the 3’ to 5’ parental DNA strand, is synthesized continuously toward the replication fork because polymerase can add nucleotides in this direction. This continuously synthesized strand is known as the leading strand. The other strand, complementary to the 5’ to 3’ parental DNA, grows away from the replication fork, so the polymerase must move back toward the replication fork to begin adding bases to a new primer, again in the direction away from the replication fork. It does so until it bumps into the previously synthesized strand and then it moves back again (Fig. 2.16). These steps produce small DNA sequence fragments known as Okazaki fragments, each separated by RNA primer. The strand with the Okazaki fragments is known as the lagging strand, and its synthesis is said to be discontinuous.
The leading strand can be extended from one primer alone, whereas the lagging strand needs a new primer for each of the short Okazaki fragments. The overall direction of the lagging strand will be 3’ to 5’, and that of the leading strand 5’ to 3’. A ring-shaped protein called the sliding clamp holds the DNA polymerase in place as it continues to add nucleotides. Beyond its role in initiation, topoisomerase prevents the overwinding of the DNA double helix ahead of the replication fork as the DNA is opening up; it does so by causing temporary nicks in the DNA helix and then resealing it. As synthesis proceeds, the RNA primers are removed by the ribonuclease activity of DNA polymerase I, which also proceeds to fill the gaps with deoxyribonucleotides. The nicks that remain between the newly synthesized DNA (that replaced the RNA primer) and the previously synthesized DNA in the adjacent Okazaki fragment are sealed by the enzyme DNA ligase that catalyzes the formation of covalent phosphodiester linkage between the 3’-OH end of one DNA fragment and the 5’ phosphate end of the other fragment, stabilizing the sugar-phosphate backbone of the DNA molecule (Fig. 2.15).
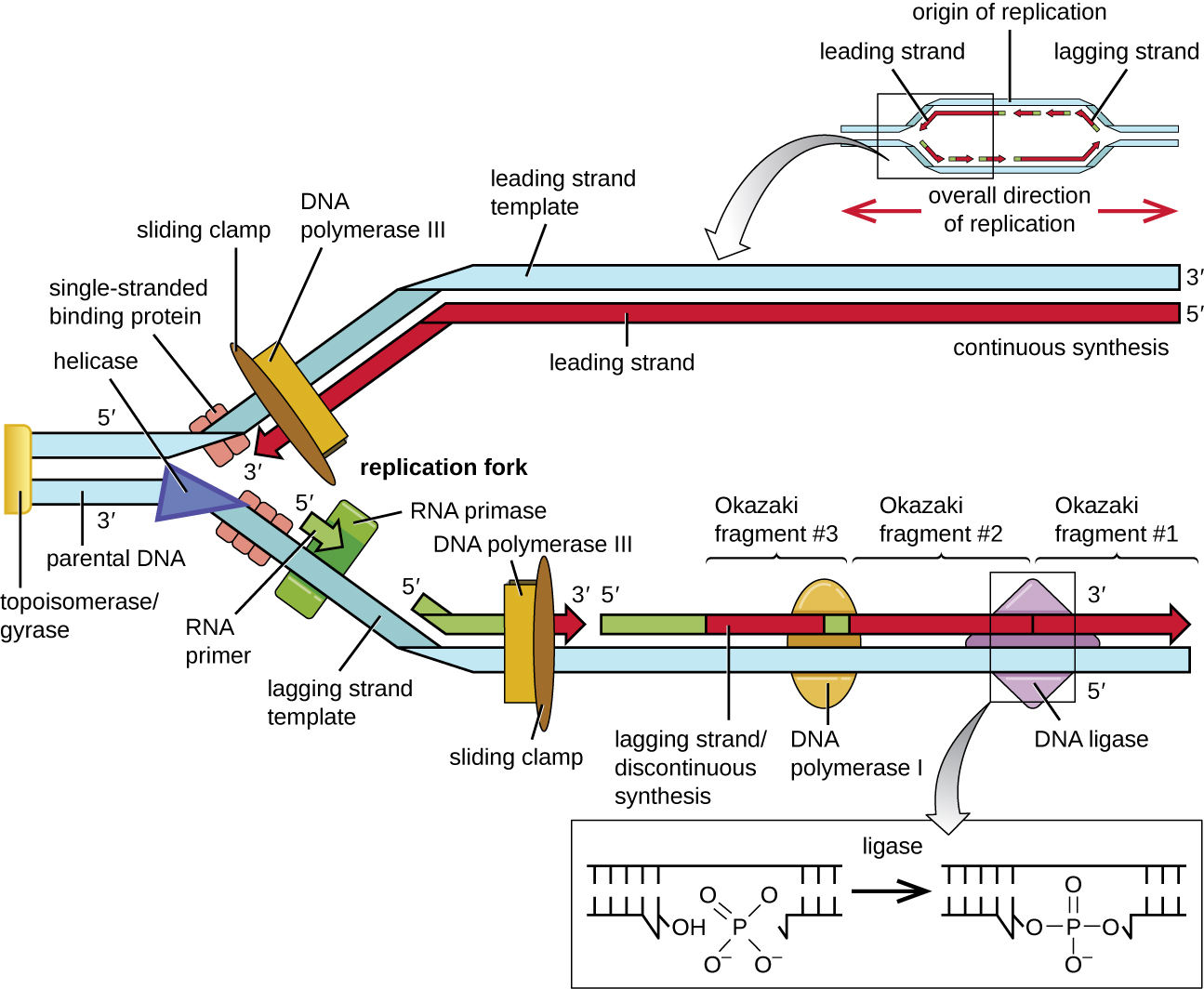
Figure 2.15. At the origin of replication, topoisomerase II relaxes the supercoiled chromosome. Two replication forks are formed by the opening of the double-stranded DNA at the origin, and helicase separates the DNA strands, which are coated by single-stranded binding proteins to keep the strands separated. DNA replication occurs in both directions. An RNA primer complementary to the parental strand is synthesized by RNA primase and is elongated by DNA polymerase III through the addition of nucleotides to the 3’-OH end. On the leading strand, DNA is synthesized continuously, whereas on the lagging strand, DNA is synthesized in short stretches called Okazaki fragments. RNA primers within the lagging strand are removed by the exonuclease activity of DNA polymerase I, and the Okazaki fragments are joined by DNA ligase. CC 4.0 by Parker.
Termination
In a region about opposite from the oriC in the chromosome is an AT-rich termination (Ter) site to which Tus (terminus utilization substance) binds. Tus blocks the advance of helicase and the entire complement of replication proteins, thus blocking progress of both replication forks and replication stops. The replicated and completed circular genomes of prokaryotes are initially concatenated and must be separated from each other. This is accomplished through the activity of topoisomerase IV, which introduces double-stranded breaks into DNA molecules, allowing them to separate from each other; the enzyme then reseals the circular chromosomes. The resolution of concatemers is unique to prokaryotic DNA replication because of their circular chromosomes. Because both bacterial DNA gyrase and topoisomerase IV are distinct from their eukaryotic counterparts, these enzymes serve as targets for a class of antimicrobial drugs called quinolones.
Table 2.2. Proteins involved in DNA replication in E. coli. The section on DNA replication is adapted from Neylon et al. and Parker.
|
Protein |
Function in DNA replication |
|
DnaA |
Initiator protein; binds to oriC |
|
DnaB |
Helicase, unwinds DNA by breaking H-binds between paired bases |
|
DnaC |
Guides DnaB to oriC |
|
SSB |
Binds to single-stranded DNA to keep DNA strands from reannealing |
|
Primase |
Synthesis of RNA primers for leading and lagging strands |
|
DNA polymerase I |
Exonuclease activity for primer removal; DNA synthesis |
|
DNA polymerase III |
Main enzyme involved in DNA synthesis; editing |
|
DNA ligase |
Seals DNA nicks by catalyzing formation of phosphodiester bonds |
|
DNA gyrase |
Topoisomerase II – Relieves supercoiling |
2.5 Regulation of chromosome replication
Cell division in both prokaryotes and eukaryotes cannot proceed without DNA replication to assure that the resulting daughter cells would both possess genomes. There are at least four conditions that need to be present for DNA replication to proceed.
- DnaA concentration. ATP-DnaA is inactivated after each round of replication to ADP-DnaA. A high number of DnaA (needs to form oligomers) is required to be bound to the DnaA boxes in oriC to promote strand separation.
- High ATP to ADP ratio. DNA replication in bacteria is mainly governed by the ATP to ADP threshold within the cell. DnaA requires ATP to melt the origin of replication, but binds equally well to both ADP and ATP. Only rapidly growing cells ready to divide will have more ATP than ADP, and therefore, initiate DNA replication. DNA-ATP is also required for the large DnaA multimer complex.
- Cell mass. Cell size correlates with cell elongation. Once newly formed cells reach the size of the parental cell, and the cell is preparing to divide, DNA is replicated as a precursor to cell division. As full cell size is reached sooner in rich media, replication also initiates sooner in rich media. DNA replication starts even before earlier round is finished.
- Methylated DNA. In the oriC, there are 11 palindromic GATC motifs where the adenines in both strands get fully methylated in fully mature cells (Fig. 2.16). DnaA binds strongest to oriC with fully methylated DNA. After a fresh round of DNA synthesis, the adenines in the newly synthesized daughter strands of DNA will remain unmethylated for some time – there is a 10-13 min delay before adenines in new strand are methylated. Thus, the double stranded oriC is only partially methylated (hemi-methylated) for some time. As DnaA binds weakly to hemi-methylated DNA, replication downregulates itself.
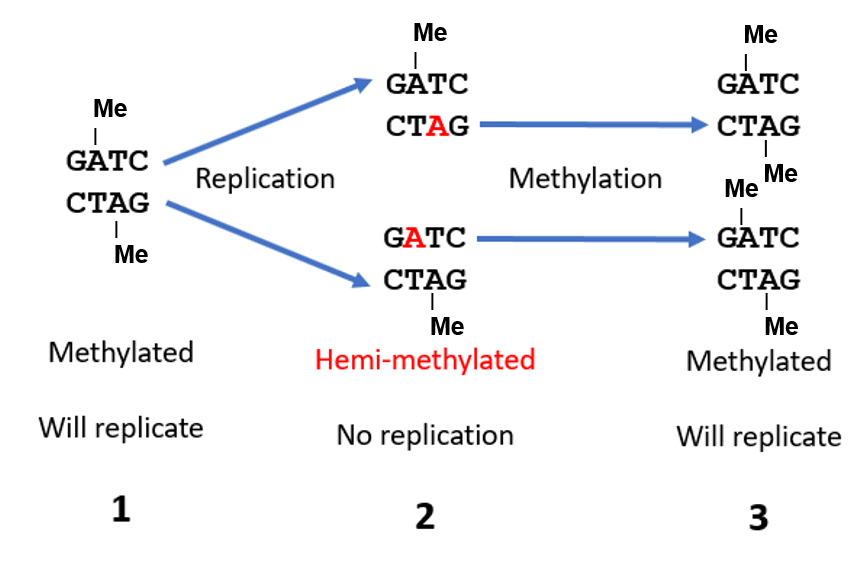
Figure 2.16. Regulation of DNA replication by methylation. DNA sequences in the oriC of mature cells are fully methylated. For some time after DNA replication and cell division, the adenines in newly synthesized DNA in daughter cells remain unmethylated (red A); the double stranded DNAs are said to be hemi-methylated. The adenines are then methylated by a DAM methylase, in which case the oriC DNA is again ready to accept DnaA to initiate the next round of DNA replication. Modified from Hardison.
2.6 Antibiotics that inhibit DNA replication
Since DNA replication is an essential function prior to cell division, it is an important target for antibiotics. Some drugs that interfere with DNA replication are:
- Trimethoprim and methotrexate inhibit nucleotide synthesis. Methotrexate is an antitumor agent and also a drug for arthritis. It has 1000X more affinity against the bacterial enzyme than to the human enzyme. Trimethoprim is bacteriostatic (as opposed to bactericidal).
- 5-Bromouracil has bromine in place of methyl group (in thymidine). It is mis-incorporated in place of thymine and increases replication errors. 5-BU is also an antitumor together with its analogue, fluorouracil.
- Ciprofloxacin is highly effective against both Gram + and Gram – bacteria. It binds to DNA gyrase (Topoisomerase II). It is a common drug for many sexually transmitted bacteria and highly effective against antibiotic resistant microbes (Superbugs).
2.7 Uses of DNA polymerases
1. Polymerase Chain Reaction. Adapted from Hartline (CC BY-NC-NA 4.0)
Polymerase chain reaction (PCR) is a technique that scientists use to amplify (make many copies of) specific DNA regions for further analysis. PCR has a huge variety of applications including:
- diagnosis of a microbial infection
- studying disease-causing organisms
- determining the presence of difficult to culture, or unculturable, microorganisms in humans or environmental samples
- amplifying a target region of DNA for cloning into a plasmid vector
- detecting genetic diseases
- cloning gene fragments to analyze genetic diseases
- identifying contaminant foreign DNA in a sample
- preparing DNA for sequencing
- determining paternity
- identifying the source of a DNA sample left at a crime scene
- comparing samples of ancient DNA with modern organisms
Most methods of DNA analysis, such as restriction enzyme digestion and agarose gel electrophoresis, or DNA sequencing require large amounts of a specific DNA fragment. In the past, large amounts of DNA were produced by growing the host cells of a genomic library. However, libraries take time and effort to prepare and DNA samples of interest often come in minute quantities. The polymerase chain reaction (PCR) permits rapid amplification in the number of copies of specific DNA sequences for further analysis. One of the most powerful techniques in molecular biology, PCR was developed in 1983 by Kary Mullis.
Taq Polymerase: An Enzyme Making PCR Possible
PCR is an in vitro laboratory technique that takes advantage of the natural process of DNA replication. Recall that DNA replication requires a DNA polymerase enzyme to build a DNA molecule complementary to a template DNA molecule. In PCR, A heat-stable DNA polymerase enzyme is used since PCR requires high temperatures to denature DNA (separate double-stranded DNA to make the DNA single-stranded). The heat-stable DNA polymerase does not denature in these conditions since it is derived from a hyperthermophilic bacterial species (“loves” hot temperatures) called Thermus aquaticus. Taq polymerase is a DNA polymerase from T. aquaticus. T. aquaticus was first isolated from a hot spring in Yellowstone National Park and thrives in very hot temperatures.
DNA Replication (and PCR) Require Primers
DNA replication requires the use of primers for the initiation of replication. Recall that DNA polymerases can only elongate a DNA molecule and cannot build a new strand from the start. In living cells, DNA replication uses the enzyme primase to build primers composed of RNA that DNA polymerase can elongate to build new DNA strands. In the laboratory setting, RNA is not very stable, and therefore, DNA primers are used for PCR. Primers not only are necessary for a DNA polymerase to elongate and produce DNA copies. Primers are used in PCR to target a specific DNA sequence that we want to copy. Primer sequences are specifically designed and engineered with a specific sequence in order to target a specific DNA region. This ensures that only the DNA sequence we want to copy gets copied and not other places in the DNA template molecule(s).
For example, if PCR is used for diagnosis of a microbial infection in a human, primers would be designed with sequences that match a specific region of DNA in that microbe and does not match DNA in other microbes or in humans. If the DNA is successfully copied (amplified) and matches positive controls (samples that contain the microbe the test is looking for), that microbe is causing infection in that human.
PCR Uses Temperature Cycles to Amplify DNA
PCR occurs over multiple cycles (Fig. 2.17). Each cycle containing three steps: denaturation, annealing, and extension. Machines called thermal cyclers are used for PCR; these machines can be programmed to automatically cycle through the temperatures required at each of the denaturation, annealing, and extension steps.
Denaturation: First, double-stranded template DNA containing the target sequence is denatured at approximately 95 °C. The high temperature required to physically separate the DNA strands by breaking the H bonds between bases is the reason the heat-stable DNA polymerase is required.
Annealing: Next, the temperature is lowered to approximately 55 °C (this can vary based on the PCR protocol and primer sequences). This allows the DNA primers complementary to the target sequence to anneal (form H-bonds) to the template strands, with one primer annealing to each strand.
Extension: Finally, the temperature is raised to 72 °C, the optimal temperature for the activity of Taq polymerase, allowing for the addition of nucleotides to the primer using the single-stranded target as a template.
Each cycle (denaturation, annealing, and extension) doubles the number of double-stranded target DNA copies. Typically, PCR protocols include 25–40 cycles, allowing for the amplification of a single target sequence by tens of millions to over a trillion.
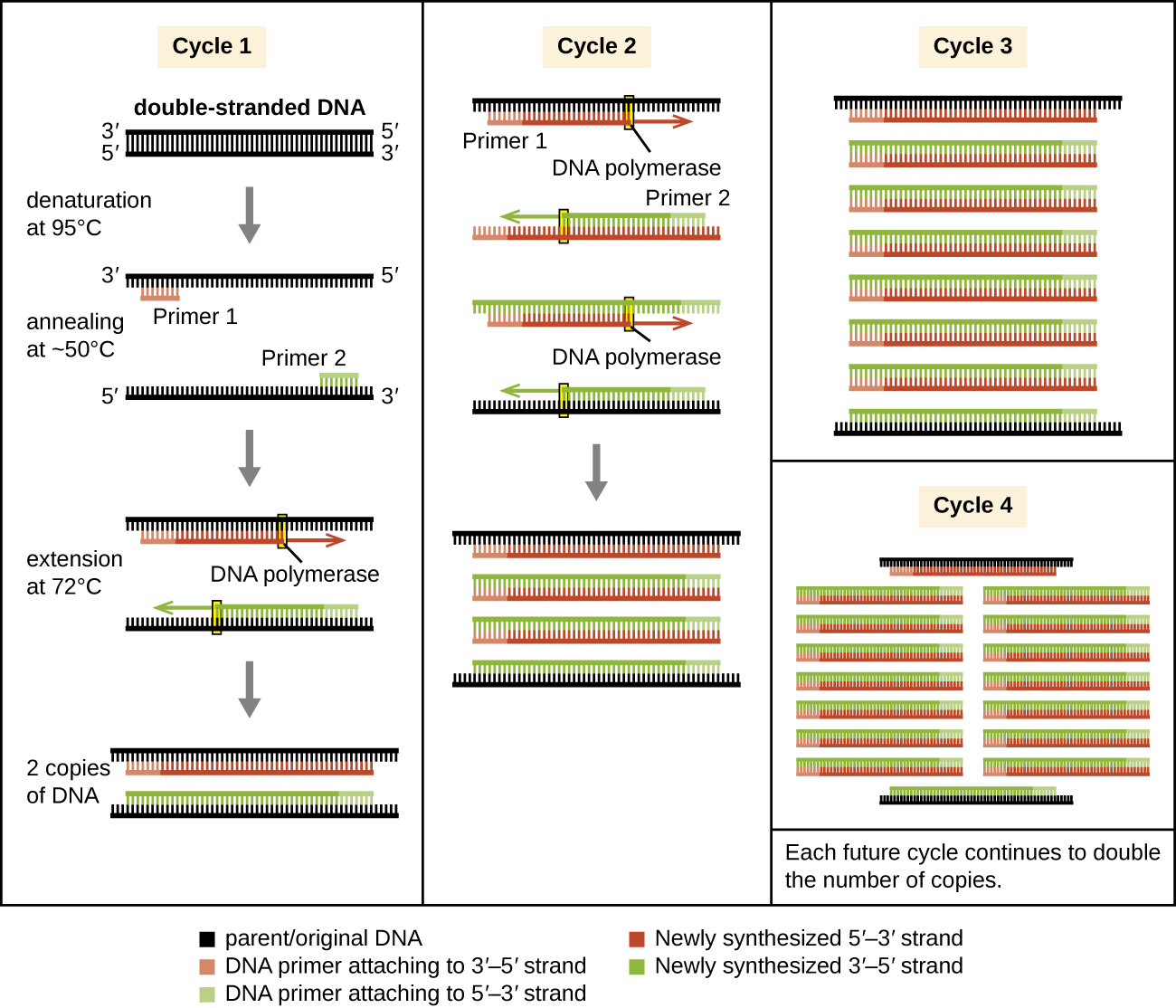
Figure 2.17.The polymerase chain reaction (PCR) is used to produce many copies of a specific sequence of target DNA. CC-SA-4.0 Parker.
Watch polymerase chain reaction :
Analysis of PCR Products
In order to analyze the PCR products after the DNA region of interest has been amplified, it is common for gel electrophoresis to be used to separate DNA based on size (Fig. 2.18). The PCR products can then be compared with controls and other samples to make conclusions about the sample.

Figure 2.18. Agarose gel electrophoresis of samples amplified by PCR. The first lane (left) contains a DNA ladder with DNA fragments of known lengths. The DNA ladder contains a variety of DNA fragment lengths to enable comparison of the DNA fragment sizes of samples with unknown lengths. Notice that there is mainly one band in each of the other four wells. This is because the DNA in the samples have been specifically targeted amplified by PCR creating millions to trillions of DNA copies of the same section of DNA. If a sample did not contain the DNA region targeted by the primers, no band would be observed. Flavier CC1-0.
2. Sanger DNA Sequencing
Until the 1990s, the sequencing of DNA was a relatively expensive and long process. Using radiolabeled nucleotides compounded the problem through safety concerns. With currently available technology and automated machines, the process is cheaper, safer, and can be completed in a matter of hours. Fred Sanger developed the sequencing method used for the human genome sequencing project, which is widely used today. The sequencing method is known as the dideoxy chain termination method (Fig. 2.19). The method is based on the use of chain terminators, the dideoxynucleotides (ddNTPs). The ddNTPSs differ from the deoxynucleotides (dNTPs) by the lack of a free 3′ OH group on the five-carbon sugar. If a ddNTP is added to a growing DNA strand, the chain cannot be extended any further because the free 3′ OH group needed to add another nucleotide is not available. By using a predetermined ratio of dNTPs to ddNTPs, it is possible to generate DNA fragments of different sizes.
The DNA sample to be sequenced is denatured (separated into two strands by heating it to high temperatures). The reaction mix consists of one sequencing primer, DNA polymerase, and all four nucleoside triphosphates (A, T, G, and C). In addition, limited quantities of the four dideoxynucleoside triphosphates (ddCTP, ddATP, ddGTP, and ddTTP) are added. For detection purposes, each of the four dideoxynucleotides carries a different fluorescent label. Chain elongation continues until a fluorescent dideoxynucleotide is incorporated, after which no further elongation takes place. After the reaction is over, capillary electrophoresis is performed to separate the fragments. Even a difference in length of a single base can be detected. The sequence is read from a laser scanner that detects the fluorescent marker of each fragment. For his work on DNA sequencing, Sanger received a Nobel Prize in Chemistry in 1980. Direct quote Adapted from Clark, Douglas and Choi (Openstax)
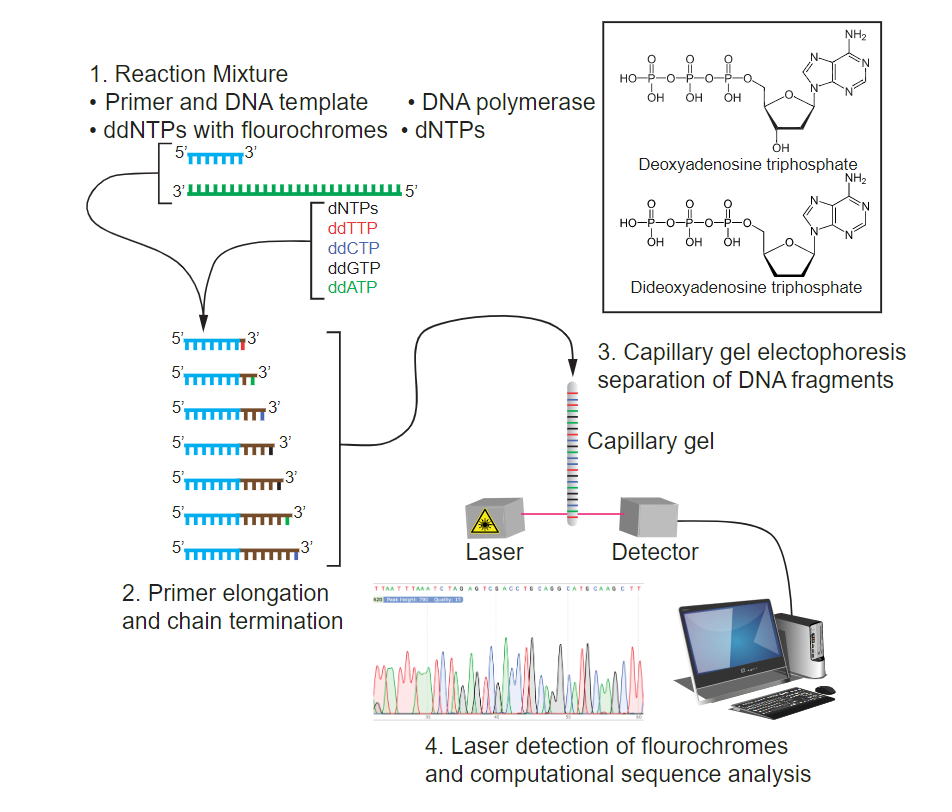
Figure 2.19. Sanger DNA sequencing. 1. The sequencing reaction consists of DNA polymerase, a sequencing primer, the four dNTPs, and four ddNTPS each labelled with four different fluorescent dyes. 2. Nucleotides get added to the 3’ end of the primers and polymerization gets terminated whenever a dideoxynucleotide gets incorporated, thereby generating fragments of different lengths. 3. The different sized fragments are separated by capillary gel electrophoresis and are hit by a laser and the emitted color read by a detector. 4. The read-out shows the four bases as ‘peaks’ of different colors. CC SA 4.0 by Tdpaustian.
Watch: Sanger sequencing
CHAPTER REVIEW QUESTIONS:

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}