
16 Chapter 16 – Applications in Medicine: Vaccines and Antibiotics
Albert B. Flavier
CHAPTER OUTLINE:
16.1 Vaccines for disease prevention
Learning Objectives
By the end of this chapter, you will be able to:
- Compare and contrast the different types of vaccines
- Describe how vaccines protect from disease
- Describe the mechanisms of action of antibiotics
- Describe how microbes develop resistance to antibiotics
Microbial pathogens have posed persistent threats to human health throughout history (Table 16.1). Biotechnology plays a significant role in the prevention, diagnosis, and treatment of diseases caused by microbial pathogens. The most effective way to avoid diseases caused by microbes is through prevention. Vaccination plays a key role in this process by using inactivated or weakened pathogens, or their components, to stimulate the body’s immune system and build resistance against infectious agents. If an infection does occur, a variety of diagnostic tools are available to detect and identify the specific microorganism responsible. Once the pathogen is accurately identified, appropriate medications, such as antibiotics for bacterial infections, can be administered to treat the disease effectively.
Table 16.1. Death toll worldwide from major pandemics.
| Disease | Death toll |
| Smallpox (to 1979) | >300,000,000 |
| Black plague (1347-1351) | 225,000,000 |
| Spanish Flu (H1N1) (1918-1919) | 50,000,000 |
| HIV/AIDS (1981-to present) | >42,300,000 (WHO) |
| Cholera (at present) | 21,000-143,000 per year |
| Covid-19 (2019-present) | >7,100,000 |
16.1 Vaccines for disease prevention
Few strategies for disease prevention are as effective as vaccination. Vaccination is a form of artificial immunity. By artificially stimulating the adaptive immune defenses, a vaccine triggers memory cell production similar to that which would occur during a primary response to exposure to a microbe. In so doing, the patient is able to mount a strong secondary response upon subsequent exposure to the pathogen—but without having to first suffer through an initial infection. Artificial active immunity is the foundation for vaccination. It involves the activation of adaptive immunity through the deliberate exposure of an individual to weakened or inactivated pathogens, or preparations consisting of key pathogen antigens.
Variolation and Vaccination
More than 300M people worldwide died from smallpox before it was eradicated by vaccination. Smallpox is a highly contagious disease caused by the variola virus and is spread through coughing or sneezing. Three out of every 10 infected people die, and many survivors are left with permanent scars over their bodies (Fig. 16.1), including their faces; some are left blind. Thousands of years ago, it was first recognized that individuals who survived a smallpox infection were immune to subsequent infections. The practice of inoculating individuals to actively protect them from smallpox appears to have originated in the 10th century in China, when the practice of variolation was described. Variolation refers to the deliberate inoculation of individuals with infectious material from scabs or pustules of smallpox victims. Infectious materials were either injected into the skin or introduced through the nasal route. The infection that developed was usually milder than naturally acquired smallpox and recovery from the milder infection provided protection against the more serious disease. Although the majority of individuals treated by variolation developed only mild infections, the practice was not without risks. More serious and sometimes fatal infections did occur, and because smallpox was contagious, infections resulting from variolation could lead to epidemics.
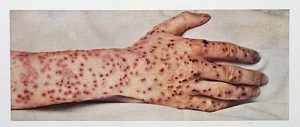
Figure 16.1. Smallpox lesions. CC-BY SA 4.0 from the Wellcome Collection (From CDC).
Although variolation had been practiced for centuries, the English physician Edward Jenner (1749–1823) is generally credited with developing the modern process of vaccination. Jenner observed that cow milkers who developed cowpox, a disease similar to smallpox but milder, were immune to the more serious smallpox. This led Jenner to hypothesize that exposure to a less virulent pathogen could provide immune protection against a more virulent pathogen, providing a safer alternative to variolation. In 1796, Jenner tested his hypothesis by obtaining infectious samples from a person’s active cowpox lesion and injecting the materials into a young boy. The boy developed a mild infection. When the boy was later infected with infectious samples from smallpox lesions, he did not contract smallpox. This new approach was termed vaccination, a name derived from the use of cowpox (Latin vacca meaning “cow”) to protect against smallpox. Today, we know that Jenner’s vaccine worked because the cowpox virus is genetically and antigenically related to the Variola viruses that caused smallpox. Exposure to cowpox antigens resulted in a primary response and the production of memory cells against identical or related epitopes of Variola virus upon a later exposure to smallpox.
Watch Jenner and the smallpox vaccine:
Because of a global smallpox vaccination program spearheaded by the World Health Organization (WHO), smallpox was declared eradicated in 1980. The success of Jenner’s smallpox vaccination led other scientists to develop vaccines for other diseases. Perhaps the most notable was Louis Pasteur, who developed vaccines for rabies, cholera, and anthrax. During the 20th and 21st centuries, effective vaccines were developed to prevent a wide range of diseases caused by viruses (e.g., chickenpox and shingles, hepatitis, measles, mumps, polio, and yellow fever) and bacteria (e.g., diphtheria, pneumococcal pneumonia, tetanus, and whooping cough).
Classes of Vaccines
For a vaccine to provide protection against a disease, it must expose an individual to pathogen-specific antigens that will stimulate a protective adaptive immune response. By its very nature, this entails some risk. As with any pharmaceutical drug, vaccines have the potential to cause adverse effects. However, the ideal vaccine causes no severe adverse effects and poses no risk of contracting the disease that it is intended to prevent. Vaccines are used not only to protect against infectious diseases but recently are being developed as treatment for various cancers. Various types of vaccines have been developed with these goals in mind (Fig. 16.2).
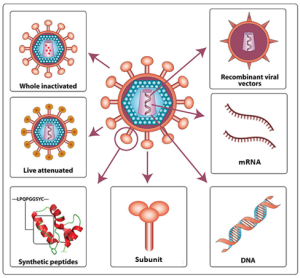
Figure 16.2. Classes of vaccines. From top left, going counterclockwise: Whole inactivated, live attenuated, synthetic peptides, subunit, DNA, mRNA and recombinant viral vectored vaccines. Modified from Bluerasberry. CC 2.0
- Live Attenuated Vaccines
Live attenuated vaccines expose an individual to a weakened strain of a pathogen with the goal of establishing a subclinical infection that will activate the adaptive immune defenses. Pathogens are attenuated to decrease their virulence using methods such as genetic manipulation (to eliminate key virulence factors) or long-term culturing in an unnatural host or environment or growth media (to promote mutations and decrease virulence).
By establishing an active infection, live attenuated vaccines stimulate a more comprehensive immune response than the other types of vaccines. Live attenuated vaccines activate both cellular and humoral immunity and stimulate the development of memory for long-lasting immunity. In some cases, vaccination of one individual with a live attenuated pathogen can even lead to natural transmission of the attenuated pathogen to other individuals. This can cause the other individuals to also develop an active, subclinical infection that activates their adaptive immune defenses.
Disadvantages associated with live attenuated vaccines include the challenges associated with long-term storage and transport as well as the potential for a patient to develop signs and symptoms of disease during the active infection (particularly in immunocompromised patients). There is also a risk of the attenuated pathogen reverting back to full virulence. Some of the more common attenuated vaccines are those the MMR, BCG for tuberculosis, nasal flu and chickenpox vaccines.
- Inactivated Vaccines
Inactivated vaccines contain whole pathogens that have been killed or inactivated with heat, chemicals, or radiation. For inactivated vaccines to be effective, the inactivation process must not affect the structure of key antigens on the pathogen.
Because the pathogen is killed or inactive, inactivated vaccines do not produce an active infection, and the resulting immune response is weaker and less comprehensive than that provoked by a live attenuated vaccine. Typically, the response involves only humoral immunity, and the pathogen cannot be transmitted to other individuals. In addition, inactivated vaccines usually require higher doses and multiple boosters, possibly causing inflammatory reactions at the site of injection.
Despite these disadvantages, inactivated vaccines do have the advantages of long-term storage stability and ease of transport. Also, there is no risk of causing severe active infections. However, inactivated vaccines are not without their side effects. Some of the more common attenuated vaccines are that for cholera, polio, rabies, and injected flu vaccines,
- Subunit Vaccines
Whereas live attenuated and inactive vaccines expose an individual to a weakened or dead pathogen, subunit vaccines only expose the patient to the key antigens of a pathogen—not whole cells or viruses. Subunit vaccines can be produced either by chemically degrading a pathogen and isolating its key antigens or by producing the antigens through recombinant DNA technology and genetic engineering of production hosts. Because these vaccines contain only the essential antigens of a pathogen, the risk of side effects is relatively low.
Subunit vaccines can be produced by expressing certain pathogen genes in heterologous host – in bacteria, yeast, insect, mammalian – and then purifying the translated protein products. Gardasil is a recombinant vaccine where subunits from the human papilloma virus (HPV) are produced using yeast. HPV causes several types of cancers including cervical cancer which was once the leading cause of cancer deaths among women in the U.S. Screening and the HPV vaccine have made cervical cancer preventable.
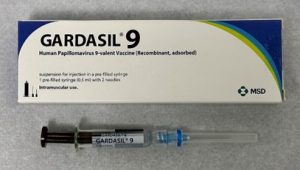
Figure 16.3. Gardasil vaccine against human papilloma virus. CC BY-SA 4.0. (From EMA)
Toxoid vaccines can be considered as a type of subunit vaccines. Toxoids consist of inactivated and purified bacterial exotoxins. Examples are the diphtheriae, tetanus and pertussis and (DTaP or TDaP) toxoid vaccines. Toxoid vaccines do not prevent bacterial infections but serve to neutralize the toxins and prevent cellular damage caused by the toxins.
- DNA Vaccines
A DNA vaccine is produced by incorporating genes for antigens into a recombinant plasmid. DNA is contained in liposomes or lipid nanoparticles (LNPs) to protect them from nucleases and facilitate delivery and fusion with cell membranes. Introduction of the DNA vaccine into a patient leads to uptake of the recombinant plasmid by some of the patient’s cells, followed by transcription and translation of antigens and presentation of these antigens with MHC I to activate adaptive immunity. This results in the stimulation of both humoral and cellular immunity without the risk of active disease associated with live attenuated vaccines.
Currently, there are no DNA vaccines approved for human use. First-generation DNA vaccines tested in the 1990s looked promising in animal models but were disappointing when tested in human subjects. Poor cellular uptake of the DNA plasmids was one of the major problems impacting their immunogenicity and efficacy.
- RNA Vaccines
mRNA vaccines were first used commercially to protect against the SARS-2 coronavirus which caused a pandemic in 2019 that killed more than a million in the US alone. The mRNA is synthesized in vitro using T7 RNA polymerase to transcribe the mRNA driven by the T7 gp10 promoter. Modified uridine derivatives are used to improve stability and reduce immunogenicity. A 5’ methyl guanosine CAP and 3’ polyA tail which also improve stability and translation efficiency are added enzymatically in vitro (Fig. 16.4). Codons are also optimized for translation in humans.
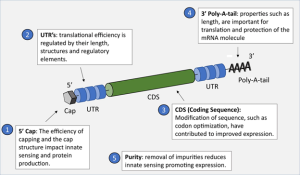
Figure 16.4. Features found on an mRNA vaccine construct. From Nicholas A. C., et al. CC-SA-4.0
Like DNA vaccines, mRNA is encapsulated or embedded in LNPs injected into patients. Unlike DNA which needs to cross the nuclear membrane for the DNA to be transcribed, mRNA once released into the cytoplasm from liposomes, is ready to be translated immediately by ribosomes (Fig. 16.5) into antigenic peptides. The peptides translated may also have the desired post-translational modifications that cannot be achieved by subunit vaccine expression in bacterial hosts.
Since mRNA can be rapidly degraded in the cytoplasm, low antigen production, and rapid decline in immunity over time, and may require high dose, scientists designed synthetic self-amplifying circular mRNA (saRNA) which incorporates an mRNA replication machinery. A SARS-2 vaccine using saRNA that requires a much lower dose was approved by the FDA in 2023 (Vallet, T. et al. 2025).
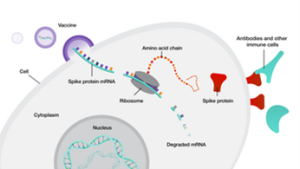
Figure 16.5. mRNA vaccine against SARS-2. mRNA is encapsulated in liposomes which fuses with the plasma membrane releasing the mRNA into the cytoplasm. The mRNA gets translated into spike protein which gets displayed on the surface of cells where they are recognized by antibodies or B cells. Image from Gonnarpax. CC SA-4.0
- Viral-vectored Vaccines
In viral-vectored vaccines (VVV), a gene from a pathogenic virus is engineered to be expressed in and be delivered by a non-pathogenic or weakly pathogenic virus that could infect but not replicate in human cells. Like attenuated and inactivated vaccines, VVVs elicit stronger T-cell responses. Ervebo is a live, attenuated recombinant virus vaccine administered to protect from the deadly Ebola virus (CDC). To make Ervebo, vesicular stomatitis virus (VSV) was genetically engineered to carry a gene from the Ebola virus that codes for a surface glycoprotein. Certain vaccines against the SARS2 virus were made by engineering the SARS2 spike protein in adenovirus, the common cold virus (Grimmett et al. 2022).
- Synthetic vaccines
Synthetic vaccines consist mainly of synthetic peptides, carbohydrates, or antigens. They are considered to be safer than vaccines from live cultures. Synthetic vaccines have been made based on the diphtheria toxin and for malaria (Wikipedia).
Once doctors diagnose an infectious agent, the microbe can be eradicated by the use of antibiotics. By identifying proteins essential for bacterial metabolism and/or virulence, scientists can also identify or develop compounds that would interfere with the proteins’ functions and compromise the pathogens’ ability to survive and multiply. From knowing the important microbial functions and the essential proteins, then we can develop drugs that may block the activity of those proteins.
The First Antimicrobial Drugs
Societies relied on traditional medicines for thousands of years. Chemical analyses of the skeletal remains of people from Nubia (now found in present-day Sudan) dating from between 350 and 550 AD have shown residue of the antimicrobial agent, tetracycline. The antimicrobial properties of certain plants may also have been recognized by various cultures around the world, including Indian and Chinese herbalists. In the early 1900s, the German physician and scientist Paul Ehrlich set out to discover or synthesize chemical compounds capable of killing infectious microbes. After screening more than 600 arsenic-containing compounds, Ehrlich’s assistant found Compound 606 which successfully cured syphilis in rabbits and soon after was marketed to treat syphilis in humans. A few decades later, German scientists Josef Klarer, Fritz Mietzsch, and Gerhard Domagk discovered the antibacterial activity of a synthetic dye, prontosil, that could treat streptococcal and staphylococcal infections in mice and later in humans. Gerhard Domagk (1895–1964) was awarded the Nobel Prize in Medicine in 1939 for his work with prontosil and sulfanilamide, the active breakdown product of prontosil in the body. Sulfanilamide, the first synthetic antimicrobial created, served as the foundation for the chemical development of a family of sulfa drugs.
In 1928, Alexander Fleming (Fig. 16.6) returned from holiday and examined some old plates of staphylococci in his research laboratory in London. He observed that contaminating mold growth (subsequently identified as a strain of Penicillium notatum) inhibited staphylococcal growth on one plate. Fleming, therefore, is credited with the discovery of penicillin, the first natural antibiotic. Further experimentation showed that penicillin from the mold was antibacterial against streptococci, meningococci, and Corynebacterium diphtheriae, the causative agent of diphtheria. Industrial production of penicillin involved media optimization as well as isolation of more vigorous growing and higher-producing Penicillium strains.

Figure 16.6. Photo of Alexander Fleming who discovered penicillin. Public domain.
Bacteriostatic Versus Bactericidal
Antibacterial drugs can be either bacteriostatic or bactericidal in their interactions with target bacteria. Bacteriostatic drugs cause a reversible inhibition of growth, with bacterial growth restarting after elimination of the drug. By contrast, bactericidal drugs kill their target bacteria. The decision of whether to use a bacteriostatic or bactericidal drugs depends on the type of infection and the immune status of the patient. In a patient with strong immune defenses, bacteriostatic and bactericidal drugs can be effective in achieving clinical cure. However, when a patient is immuno-compromised, a bactericidal drug is essential for the successful treatment of infections. Regardless of the immune status of the patient, life-threatening infections such as acute endocarditis require the use of a bactericidal drug.
Spectrum of Activity
The spectrum of activity of an antibacterial drug relates to diversity of targeted bacteria. A narrow-spectrum antimicrobial targets only specific subsets of bacterial pathogens. For example, some narrow-spectrum drugs only target gram-positive bacteria, whereas others target only gram-negative bacteria. If the pathogen causing an infection has been identified, it is best to use a narrow-spectrum antimicrobial and minimize collateral damage to the normal microbiota. A broad-spectrum antimicrobial targets a wide variety of bacterial pathogens, including both gram-positive and gram-negative species, and is frequently used as empiric therapy to cover a wide range of potential pathogens while waiting on the laboratory identification of the infecting pathogen. Broad-spectrum antimicrobials are also used for polymicrobic infections (mixed infection with multiple bacterial species), or as prophylactic prevention of infections with surgery/invasive procedures. Finally, broad-spectrum antimicrobials may be selected to treat an infection when a narrow-spectrum drug fails because of development of drug resistance by the target pathogen.
The risk associated with using broad-spectrum antimicrobials is that they will also target a broad spectrum of the normal microbiota, increasing the risk of a superinfection, a secondary infection in a patient having a preexisting infection. A superinfection develops when the antibacterial intended for the preexisting infection kills the protective microbiota, allowing another pathogen resistant to the antibacterial to proliferate and cause a secondary infection. Common examples of superinfections that develop as a result of antimicrobial usage include yeast infections (candidiasis) and pseudomembranous colitis caused by Clostridioides difficile, which can be fatal.
Mechanism of action of antimicrobial drugs
An important quality for an antimicrobial drug is selective toxicity, meaning that it selectively kills or inhibits the growth of microbial targets while causing minimal or no harm to the host. Most antimicrobial drugs currently in clinical use are antibacterial because the prokaryotic cell provides a greater variety of unique targets for selective toxicity, in comparison to fungi, parasites, and viruses. Each class of antibacterial drugs has a unique mode of action (the way in which a drug affects microbes at the cellular level) and these are summarized in Figure 16.7.
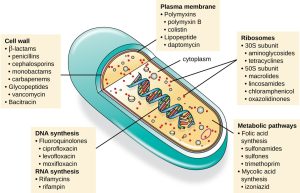
Figure 16.7. There are several classes of antibacterial compounds that are typically classified based on their bacterial target. Parker.
Cell Wall Biosynthesis Inhibitors
Several different classes of antibacterials block steps in the biosynthesis of peptidoglycan, making cells more susceptible to osmotic lysis (Table 16.1). Therefore, antibacterials that target cell wall biosynthesis are bactericidal in their action.
Penicillin is one of several antibacterials within a class called β-lactams. This group of compounds includes the penicillins, cephalosporins, monobactams, and carbapenems, and is characterized by the presence of a β-lactam ring found within the central structure of the drug molecule. The β-lactam antibacterials block the crosslinking of peptide chains during the biosynthesis of new peptidoglycan in the bacterial cell wall. They are able to block this process because the β-lactam structure is similar to the structure of the peptidoglycan subunit component that is recognized by the crosslinking transpeptidase enzyme, also known as a penicillin-binding protein (PBP). Although the β-lactam ring must remain unchanged for these drugs to retain their antibacterial activity, strategic chemical changes to the R groups have allowed for development of a wide variety of semisynthetic β-lactam drugs with increased potency, expanded spectrum of activity, and longer half-lives for better dosing, among other characteristics. Methicillin is a semisynthetic penicillin that was developed to address the spread of enzymes (penicillinases) that were inactivating the other penicillins.
Similar to the penicillins, cephalosporins contain a β-lactam ring and block the transpeptidase activity of penicillin-binding proteins. However, the β-lactam ring of cephalosporins is fused to a six-member ring, rather than the five-member ring found in penicillins. This chemical difference provides cephalosporins with an increased resistance to enzymatic inactivation by β-lactamases.
The drug bacitracin consists of a group of structurally similar peptide antibiotics originally isolated from Bacillus subtilis. Bacitracin blocks the activity of a specific cell-membrane molecule that is responsible for the movement of peptidoglycan precursors from the cytoplasm to the exterior of the cell, ultimately preventing their incorporation into the cell wall. It is combined with neomycin and polymyxin in topical ointments such as Neosporin.
Inhibitors of Membrane Function
The polymyxins are lipophilic with detergent-like properties and interact with the lipopolysaccharide component of the outer membrane of gram-negative bacteria, ultimately disrupting both their outer and inner membranes and killing the bacterial cells. Unfortunately, the membrane-targeting mechanism is not a selective toxicity, and these drugs also target and damage the membrane of cells in the kidney and nervous system when administered systemically. Because of these serious side effects and their poor absorption from the digestive tract, polymyxin B is used in over-the-counter topical antibiotic ointments (e.g., Neosporin),
Nucleic Acid Synthesis Inhibitors
Nalidixic acid selectively inhibits the activity of bacterial DNA gyrase, blocking DNA replication. The nalidixic acid derivative, ciprofloxacin, is effective against a broad spectrum of gram-positive or gram-negative bacteria, and is among the most commonly prescribed antibiotics used to treat a wide range of infections, including urinary tract infections, respiratory infections, abdominal infections, and skin infections. However, despite their selective toxicity against DNA gyrase, side effects include phototoxicity, neurotoxicity, cardiotoxicity, glucose metabolism dysfunction, and increased risk for tendon rupture.
The drug rifampin is semisynthetic and functions by blocking RNA polymerase activity in bacteria. Rifampin binds inside the active channel of RNA polymerase thus preventing the elongation of RNA transcript and halting the polymerase at the promoter. The RNA polymerase enzymes in bacteria are structurally different from those in eukaryotes, providing for selective toxicity against bacterial cells. Despite the selectivity of its mechanism, rifampin can induce liver enzymes to increase metabolism of other drugs being administered (antagonism), leading to hepatotoxicity (liver toxicity) and negatively influencing the bioavailability and therapeutic effect of the companion drugs. In recombinant protein expression, rifampin can be added to inhibit E. coli RNA polymerase while T7 RNA polymerase activated promoters are unaffected and are preferentially expressed.
Actinomycins, produced by Streptomyces parvulus, intercalates in DNA in the transcription bubble to interfere with RNA elongation. It is used more for cancer treatment rather than as an antibiotic because of its non-specific effect on RNA polymerase
Other drugs such as sulfonamides, trimethoprim and methotrexate inhibit enzymes involved in nucleotide synthesis thus interfering with DNA and RNA synthesis.
Protein Biosynthesis Inhibitors
The cytoplasmic ribosomes found in animal cells (80S) are structurally distinct from those found in bacterial cells (70S), making protein biosynthesis a good selective target for antibacterial drugs.
Aminoglycosides are large, highly polar antibacterial drugs that bind to the 30S subunit of bacterial ribosomes, impairing the proofreading ability of the ribosomal complex. This impairment causes mismatches between codons and anticodons, resulting in the production of proteins with incorrect amino acids and shortened proteins that insert into the cytoplasmic membrane. Disruption of the cytoplasmic membrane by the faulty proteins kills the bacterial cells. The aminoglycosides, which include drugs such as streptomycin, gentamicin, neomycin, and kanamycin, are potent broad-spectrum antibacterials. However, aminoglycosides have been shown to be nephrotoxic (damaging to kidney), neurotoxic (damaging to the nervous system), and ototoxic (damaging to the ear).
Another class of antibacterial compounds that bind to the 30S subunit is the tetracyclines. In contrast to aminoglycosides, these drugs are bacteriostatic and inhibit protein synthesis by blocking the association of tRNAs with the ribosome during translation. Although the tetracyclines are broad spectrum in their coverage of bacterial pathogens, side effects that can limit their use include phototoxicity, permanent discoloration of developing teeth, and liver toxicity with high doses or in patients with kidney impairment.
There are several classes of antibacterial drugs that work through binding to the 50S subunit of bacterial ribosomes. The macrolide antibacterial drugs have a large, complex ring structure and are part of a larger class of naturally produced secondary metabolites called polyketides, complex compounds produced in a stepwise fashion through the repeated addition of two-carbon units by a mechanism similar to that used for fatty acid synthesis. Macrolides are broad-spectrum, bacteriostatic drugs that block elongation of proteins by inhibiting peptide bond formation between specific combinations of amino acids. The first macrolide was erythromycin. It was from Streptomyces erythreus and prevents translocation.
Chloramphenicol, produced by Streptomyces venezuelae, is the first broad-spectrum antibiotic that was approved by the FDA. As a result of its mass production, broad-spectrum coverage, and ability to penetrate into tissues efficiently, chloramphenicol was historically used to treat a wide range of infections, from meningitis to typhoid fever to conjunctivitis. Unfortunately, serious side effects, such as lethal gray baby syndrome, and suppression of bone marrow production, have limited its clinical role. Chloramphenicol also causes anemia by targeting of mitochondrial ribosomes within hematopoietic stem cells, causing suppression of blood cell production. Because of toxicity concerns, chloramphenicol usage in humans is now rare in the United States and is limited to severe infections unable to be treated by less toxic antibiotics. Because its side effects are much less severe in animals, it is used in veterinary medicine.
Inhibitors of Metabolic Pathways
Some synthetic drugs control bacterial infections by functioning as antimetabolites, competitive inhibitors for bacterial metabolic enzymes. The sulfonamides (sulfa drugs) are the oldest synthetic antibacterial agents and are structural analogues of para-aminobenzoic acid (PABA), an early intermediate in folic acid synthesis which is required for synthesis of nucleotides. Because humans obtain folic acid from food instead of synthesizing it intracellularly, sulfonamides are selectively toxic for bacteria. However, allergic reactions to sulfa drugs are common.
Trimethoprim is a synthetic antimicrobial compound that serves as an antimetabolite within the same folic acid synthesis pathway as sulfonamides. However, trimethoprim is a structural analogue of dihydrofolic acid and inhibits a later step in the metabolic pathway. Trimethoprim is used in combination with the sulfa drug sulfamethoxazole to treat urinary tract infections, ear infections, and bronchitis. As discussed, the combination of trimethoprim and sulfamethoxazole is an example of antibacterial synergy. When used alone, each antimetabolite only decreases production of folic acid to a level where bacteriostatic inhibition of growth occurs. However, when used in combination, inhibition of both steps in the metabolic pathway decreases folic acid synthesis to a level that is lethal to the bacterial cell. Because of the importance of folic acid during fetal development, sulfa drugs and trimethoprim use should be carefully considered during early pregnancy.
16.3 Antimicrobial resistance
Antimicrobial resistance is not a new phenomenon. In nature, microbes are constantly evolving in order to overcome the antimicrobial compounds produced by other microorganisms. Human development of antimicrobial drugs and their widespread clinical use have simply provided another selective pressure that promotes further evolution. Several important factors can accelerate the evolution of drug resistance. These include the overuse and misuse of antimicrobials, inappropriate use of antimicrobials, subtherapeutic dosing, and patient noncompliance with the recommended course of treatment. In farming animals, antibiotics are also added extensively to feeds to enhance feed utilization efficiency and weight gain especially in younger animals. It has been found that swine farmers are more likely to carry multidrug-resistant Staphylococcus aureus.
Exposure of a pathogen to an antimicrobial compound can select for chromosomal mutations conferring resistance, which can be transferred vertically to subsequent microbial generations and eventually become predominant in a microbial population that is repeatedly exposed to the antimicrobial. Alternatively, many genes responsible for drug resistance are found on plasmids or in transposons that can be transferred easily between microbes through horizontal gene transfer. Transposons also have the ability to move resistance genes between plasmids and chromosomes to further promote the spread of resistance.
Mechanisms for Drug Resistance
There are several common mechanisms for drug resistance, which are summarized in Fig. 16.8. These mechanisms include enzymatic modification of the drug, modification of the antimicrobial target, and prevention of drug penetration or accumulation.
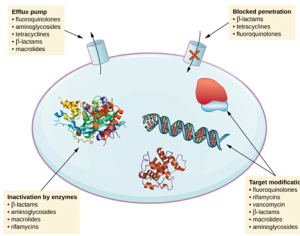
Figure 16.8. There are multiple strategies that microbes use to develop resistance to antimicrobial drugs. (credit: modification of work by Gerard D Wright). Parker
Watch Animation of Antimicrobial Resistance:
- Drug Modification or Inactivation
Resistance genes may code for enzymes that chemically modify an antimicrobial, thereby inactivating it, or destroy an antimicrobial through hydrolysis. Resistance to many types of antimicrobials occurs through this mechanism. For example, aminoglycoside resistance can occur through enzymatic transfer of chemical groups to the drug molecule, impairing the binding of the drug to its bacterial target. For β-lactams, bacterial resistance can involve the enzymatic hydrolysis of the β-lactam bond within the drug molecule. Once the β-lactam bond is broken, the drug loses its antibacterial activity. This mechanism of resistance is mediated by β-lactamases, which are the most common mechanism of β-lactam resistance. Inactivation of rifampin commonly occurs through glycosylation, phosphorylation, or adenosine diphosphate (ADP) ribosylation, and resistance to macrolides and lincosamides can also occur due to enzymatic inactivation of the drug or modification.
- Prevention of Cellular Uptake or Efflux
Microbes may develop resistance mechanisms that involve inhibiting the accumulation of an antimicrobial drug, which then prevents the drug from reaching its cellular target. This strategy is common among gram-negative pathogens and can involve changes in outer membrane lipid composition, porin channel selectivity, and/or porin channel concentrations. For example, a common mechanism of carbapenem resistance among Pseudomonas aeruginosa is to decrease the amount of its OprD porin, which is the primary portal of entry for carbapenems through the outer membrane of this pathogen. Additionally, many gram-positive and gram-negative pathogenic bacteria produce efflux pumps that actively transport an antimicrobial drug out of the cell and prevent the accumulation of drug to a level that would be antibacterial. For example, resistance to β-lactams, tetracyclines, and fluoroquinolones commonly occurs through active efflux out of the cell, and it is rather common for a single efflux pump to have the ability to translocate multiple types of antimicrobials.
- Target Modification
Because antimicrobial drugs have very specific targets, structural changes to those targets can prevent drug binding, rendering the drug ineffective. Through spontaneous mutations in the genes encoding antibacterial drug targets, bacteria have an evolutionary advantage that allows them to develop resistance to drugs. This mechanism of resistance development is quite common. Genetic changes impacting the active site of penicillin-binding proteins (PBPs) can inhibit the binding of β-lactam drugs and provide resistance to multiple drugs within this class. This mechanism is very common among strains of Streptococcus pneumoniae, which alter their own PBPs through genetic mechanisms. In contrast, strains of Staphylococcus aureus develop resistance to methicillin (MRSA) through the acquisition of a new low-affinity PBP, rather than structurally alter their existing PBPs. Not only does this new low-affinity PBP provide resistance to methicillin but it provides resistance to virtually all β-lactam drugs, with the exception of the newer fifth-generation cephalosporins designed specifically to kill MRSA. Other examples of this resistance strategy include alterations in ribosome subunits, providing resistance to macrolides, tetracyclines, and aminoglycosides; lipopolysaccharide (LPS) structure, providing resistance to polymyxins; RNA polymerase, providing resistance to rifampin; DNA gyrase, providing resistance to fluoroquinolones; metabolic enzymes, providing resistance to sulfa drugs, sulfones, and trimethoprim; and peptidoglycan subunit peptide chains, providing resistance to glycopeptides.
- Target Overproduction or Enzymatic Bypass
When an antimicrobial drug functions as an antimetabolite, targeting a specific enzyme to inhibit its activity, there are additional ways that microbial resistance may occur. First, the microbe may overproduce the target enzyme such that there is a sufficient amount of antimicrobial-free enzyme to carry out the proper enzymatic reaction. Second, the bacterial cell may develop a bypass that circumvents the need for the functional target enzyme. Both of these strategies have been found as mechanisms of sulfonamide resistance. Vancomycin resistance among S. aureus has been shown to involve the decreased cross-linkage of peptide chains in the bacterial cell wall, which provides an increase in targets for vancomycin to bind to in the outer cell wall. Increased binding of vancomycin in the outer cell wall provides a blockage that prevents free drug molecules from penetrating to where they can block new cell wall synthesis.
- Target Mimicry
A recently discovered mechanism of resistance called target mimicry involves the production of proteins that prevent drugs from binding to their bacterial cellular targets. For example, fluoroquinolone resistance by Mycobacterium tuberculosis can involve the production of a protein that resembles DNA. This protein is called MfpA (Mycobacterium fluoroquinolone resistance protein A). The mimicry of DNA by MfpA results in DNA gyrase binding to MfpA, preventing the binding of fluoroquinolones to DNA gyrase.
Multidrug-Resistant Microbes and Cross Resistance
From a clinical perspective, our greatest concerns are multidrug-resistant microbes (MDRs) and cross resistance. MDRs are colloquially known as “superbugs” and carry one or more resistance mechanism(s), making them resistant to multiple antimicrobials. In cross-resistance, a single resistance mechanism confers resistance to multiple antimicrobial drugs. For example, having an efflux pump that can export multiple antimicrobial drugs is a common way for microbes to be resistant to multiple drugs by using a single resistance mechanism.
Superbugs are responsible for killing at least 1.27 million people worldwide and associated with the deaths of many millions more (FDA) in 2019. Several of the superbugs discussed in the following sections have been dubbed the ESKAPE pathogens. This acronym refers to the names of the pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.) but it is also fitting in that these pathogens are able to “escape” many conventional forms of antimicrobial therapy. As such, infections by ESKAPE pathogens can be difficult to treat and they cause a large number of nosocomial infections (acquired in healthcare settings).
Methicillin-Resistant Staphylococcus aureus (MRSA)
Methicillin, a semisynthetic penicillin, was designed to resist inactivation by β-lactamases. Unfortunately, soon after the introduction of methicillin to clinical practice, methicillin-resistant strains of S. aureus appeared and started to spread. The mechanism of resistance, acquisition of a new low-affinity PBP, provided S. aureus with resistance to all available β-lactams. Strains of methicillin-resistant S. aureus (MRSA) are widespread opportunistic pathogens and a particular concern for skin and other wound infections, but may also cause pneumonia and septicemia. Although originally a problem in health-care settings (hospital-acquired MRSA or HA-MRSA), MRSA infections are now also acquired through contact with contaminated members of the general public, called community-associated MRSA (CA-MRSA). Approximately one-third of the population carries S. aureus as a member of their normal nasal microbiota without illness, and about 6% of these strains are methicillin resistant.
This chapter is shared under a CC BY 4.0 license and was authored, remixed, and/or curated by Parker et al., at OpenStax via source content
References:
Grimmett E, Al-Share B, Alkassab MB, Zhou RW, Desai A, Rahim MMA, Woldie I. Cancer vaccines: past, present and future; a review article. Discov Oncol. 2022 May 16;13(1):31. doi: 10.1007/s12672-022-00491-4. PMID: 35576080; PMCID: PMC9108694.
Vallet T, Vignuzzi M. Self-Amplifying RNA: Advantages and Challenges of a Versatile Platform for Vaccine Development. Viruses. 2025 Apr 14;17(4):566. doi: 10.3390/v17040566. PMID: 40285008; PMCID: PMC12031284.
PRACTICE QUESTIONS:

{kind=link}
{kind=link}
{kind=link}