
6 Chapter 6. Mutations, Mutagenesis, and DNA Repair
Albert B. Flavier
Chapter Outline
Learning Objectives
By the end of this chapter, you will be able to:
Describe different types of mutations
Describe the effects of mutations on protein structure, function, and expression
Describe several approaches to mutagenesis
Describe DNA repair processes
Mutations
A mutation is a heritable change in the DNA sequence of an organism. The resulting organism, called a mutant, may have a recognizable change in phenotype compared to the wild type, where the wild type is the phenotype most commonly observed in nature. A change in the DNA sequence is conferred to mRNA through transcription and may lead to an altered amino acid sequence in a protein upon translation. Because proteins carry out the vast majority of cellular functions, a change in amino acid sequence in a protein may lead to an altered phenotype at the cellular and organismal level.
Effects of Mutations on DNA Sequence
There are several types of mutations that are classified according to how the DNA molecule is altered. One type, called a point mutation, affects a single base and most commonly occurs when one base is substituted or replaced by another. A change from a purine to another purine or pyrimidine to another pyrimidine are transition mutations, while a change from a purine to a pyrimidine or vice versa is a transversion mutation. Mutations also result from the addition of one or more bases, known as an insertion, or the removal of one or more bases, known as a deletion.
6.1 Effects of Mutations on Protein Structure and Function
Point mutations, when they occur in protein coding regions, may have a wide range of effects on protein function (Fig. 6.1). As a consequence of the degeneracy of the genetic code, a point mutation will commonly result in the same amino acid being incorporated into the resulting polypeptide despite the sequence change. This change would have no effect on the protein’s structure and is thus called a silent mutation. A missense mutation results in a different amino acid being incorporated into the resulting polypeptide. The effect of a missense mutation depends on how chemically different the new amino acid is from the wild-type amino acid. The location of the changed amino acid within the protein also is important. For example, if the changed amino acid is part of the enzyme’s active site, then the effect of the missense mutation may be significant. A mutation that leads to a protein’s complete loss of function is considered as a null mutation. Many missense mutations result in proteins that are still functional, at least to some degree, and are thus considered as leaky mutations. Sometimes the effects of missense mutations may be only apparent under certain environmental conditions; such missense mutations are called conditional mutations. A missense mutation may be beneficial. Under the right environmental conditions, this type of mutation may give the organism that harbors it a selective advantage. Yet another type of point mutation, called a nonsense mutation, converts a codon encoding an amino acid (a sense codon) into a stop codon (a nonsense codon). Nonsense mutations result in the synthesis of proteins that are shorter than the wild type and typically not functional depending largely on the length of the missing sequence.
Deletions and insertions also cause various effects. Because codons are triplets of nucleotides, insertions or deletions in groups of three nucleotides may lead to the insertion or deletion of one or more amino acids and may not cause significant effects on the resulting protein’s functionality. However, frameshift mutations, caused by insertions or deletions of a number of nucleotides that are not a multiple of three are extremely problematic because a shift in the reading frame results (Fig. 6.1). Because ribosomes read the mRNA in triplet codons, frameshift mutations can change every amino acid after the point of the mutation. The new reading frame may also include a stop codon before the end of the coding sequence. Consequently, proteins made from genes containing frameshift mutations are nearly always nonfunctional.
Mutations in Regulatory regions
Mutations can also occur outside of the protein coding sequences. Mutations in transcriptional promoter regions, enhancers or operators, can lead to downregulation or upregulation of transcription. A mutation in the ribosome binding site could also alter the rates of translation. Most mutations in regulatory sequences tend to be leaky types and can have significant effects on phenotype.
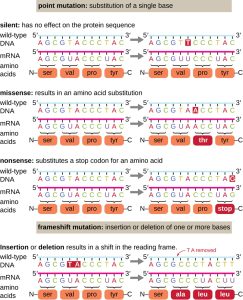
Figure 6.1. Mutations can lead to changes in the protein sequence encoded by the DNA. CC BY-SA 4.0 from Parker
6.2 Causes of Mutations
Mistakes in the process of DNA replication can cause spontaneous mutations to occur. The error rate of DNA polymerase is one incorrect base per billion base pairs replicated. Exposure to mutagens can cause induced mutations, which are various types of chemical agents or radiation (Table 6.1). Exposure to a mutagen can increase the rate of mutation more than 1000-fold. Mutagens are often also carcinogens, agents that cause cancer. However, whereas nearly all carcinogens are mutagenic, not all mutagens are necessarily carcinogens.
Chemical Mutagens
Various types of chemical mutagens interact directly with DNA either by acting as nucleoside analogs or by modifying nucleotide bases. Chemicals called nucleoside analogs are structurally similar to normal nucleotide bases and can be incorporated into DNA during replication (Fig. 6.2). These base analogs induce mutations because they often have different base-pairing rules than the bases they replace. Other chemical mutagens can modify normal DNA bases, resulting in different base-pairing rules. For example, hydroxylamine and nitrous acid deaminates cytosine, converting it to uracil. Uracil then pairs with adenine in a subsequent round of replication, resulting in the conversion of a GC base pair to an AT base pair. Nitrous acid also deaminates adenine to hypoxanthine, which base pairs with cytosine instead of thymine, resulting in the conversion of a TA base pair to a CG base pair.
Chemical mutagens known as intercalating agents work differently. These molecules slide between the stacked nitrogenous bases of the DNA double helix, distorting the molecule and creating atypical spacing between nucleotide base pairs (Fig. 6.3). As a result, during DNA replication, DNA polymerase may either skip replicating several nucleotides (creating a deletion) or insert extra nucleotides (creating an insertion). Either outcome may lead to a frameshift mutation. Combustion products like polycyclic aromatic hydrocarbons are particularly dangerous intercalating agents that can lead to mutation-caused cancers. The intercalating agents, ethidium bromide and acridine orange, are commonly used in the laboratory to stain DNA for visualization and are potential mutagens, although the mutagenic potential of ethidium bromide has been questioned.
Another class mutagens, like the commonly used ethyl methanesulfonate, are alkylating agents that add alkyl groups to guanine, leading to mispairing with thymine.
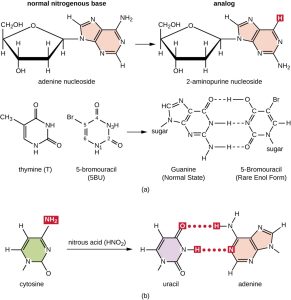
Figure 6.2. (a) 2-aminopurine nucleoside (2AP) structurally is a nucleoside analog to adenine nucleoside, whereas 5-bromouracil (5BU) is a nucleoside analog to thymine nucleoside. 2AP base pairs with C, converting an AT base pair to a GC base pair after several rounds of replication. 5BU pairs with G, converting an AT base pair to a GC base pair after several rounds of replication. (b) Nitrous acid is a different type of chemical mutagen that modifies already existing nucleoside bases like C to produce U, which base pairs with A. This chemical modification, as shown here, results in converting a CG base pair to a TA base pair. CC BY-SA 4.0 from Parker
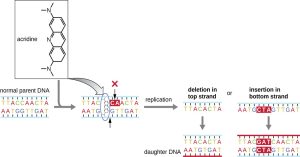
Figure 6.3. Intercalating agents, such as acridine, introduce atypical spacing between base pairs, resulting in DNA polymerase introducing either a deletion or an insertion, leading to a potential frameshift mutation. CC BY-SA 4.0 from Parker
Radiation
Exposure to either ionizing or nonionizing radiation can each induce mutations in DNA, although by different mechanisms. Strong ionizing radiation like X-rays and gamma rays can cause single- and double-stranded breaks in the DNA backbone through the formation of hydroxyl radicals on radiation exposure (Fig. 6.4). Ionizing radiation can also modify bases; for example, the deamination of cytosine to uracil, analogous to the action of nitrous acid (1). Ionizing radiation exposure is used to kill microbes to sterilize medical devices and foods, because of its dramatic nonspecific effect in damaging DNA, proteins, and other cellular components (see Using Physical Methods to Control Microorganisms).
Nonionizing radiation, like ultraviolet light, is not energetic enough to initiate these types of chemical changes. However, nonionizing radiation can induce dimer formation between two adjacent pyrimidine bases, commonly two thymines, within a nucleotide strand. During thymine dimer formation, the two adjacent thymines become covalently linked and, if left unrepaired, both DNA replication and transcription are stalled at this point. DNA polymerase may proceed and replicate the dimer incorrectly, potentially leading to frameshift or point mutations.
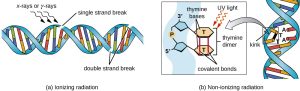
Figure 6.4. (a) Ionizing radiation may lead to the formation of single-stranded and double-stranded breaks in the sugar-phosphate backbone of DNA, as well as to the modification of bases (not shown). (b) Nonionizing radiation like ultraviolet light can lead to the formation of thymine dimers, which can stall replication and transcription and introduce frameshift or point mutations. CC BY-SA 4.0 from Parker
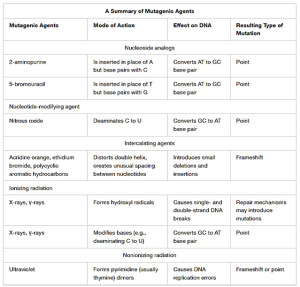
Table 6.1. A summary of mutagenic agents, their mode of action, effect on DNA and the resulting type of mutation. CC BY-SA 4.0 from Parker
6.3 DNA Repair
The process of DNA replication is more than 99.99% accurate, but mistakes can occur spontaneously or be induced by mutagens. Uncorrected mistakes can lead to serious consequences for the phenotype. Cells have developed several repair mechanisms to minimize the number of mutations that persist.
Proofreading
Most of the mistakes introduced during DNA replication are promptly corrected by most DNA polymerases through a function called proofreading. In proofreading, the DNA polymerase reads the newly added base, ensuring that it is complementary to the corresponding base in the template strand before adding the next one. If an incorrect base has been added, the enzyme makes a cut to release the wrong nucleotide, and the correct base is added.
Methyl-directed Mismatch Repair
Some errors introduced during replication are corrected shortly after the replication machinery has moved. This mechanism is called mismatch repair. The enzymes involved in this mechanism recognize the incorrectly added nucleotide, excise it, and replace it with the correct base. One example is the methyl-directed mismatch repair in E. coli. The DNA is hemimethylated. This means that the parental strand is methylated while the newly synthesized daughter strand is not. It takes several minutes before the new strand is methylated. Proteins MutS, MutL, and MutH bind to the hemimethylated site where the incorrect nucleotide is found. MutH cuts the nonmethylated strand (the new strand). An exonuclease removes a portion of the strand (including the incorrect nucleotide). The gap formed is then filled in by DNA pol III and ligase.
Repair of Pyrimidine Dimers
Because the production of pyrimidine dimers is common due to exposure to ultraviolet light, mechanisms have evolved to repair these lesions. In nucleotide excision repair (also called dark repair), enzymes remove the pyrimidine dimer and replace it with the correct nucleotides (Fig. 6.5). In E. coli, the DNA is scanned by an enzyme complex (UvrABC). If a distortion in the double helix is found that was introduced by the pyrimidine dimer, the enzyme complex cuts the sugar-phosphate backbone several bases upstream and downstream of the dimer, and the segment of DNA between these two cuts is then enzymatically removed. DNA pol I replaces the missing nucleotides with the correct ones and DNA ligase seals the gap in the sugar-phosphate backbone.
The direct repair (also called light repair) of thymine dimers occurs through the process of photoreactivation in the presence of visible light. An enzyme called photolyase and recognizes the distortion in the DNA helix caused by the thymine dimer and binds to the dimer. In the presence of visible light, the photolyase enzyme changes conformation and breaks apart the thymine dimer, allowing the thymines to again correctly base pair with the adenines on the complementary strand. Photoreactivation appears to be present in all organisms, with the exception of placental mammals, including humans. Photoreactivation is particularly important for organisms chronically exposed to ultraviolet radiation, like plants, photosynthetic bacteria, algae, and corals, to prevent the accumulation of mutations caused by thymine dimer formation.
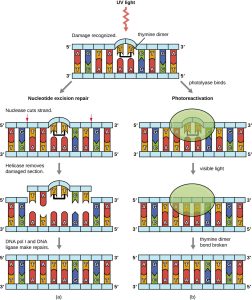
Figure 6.5. Bacteria have two mechanisms for repairing thymine dimers. (a) In nucleotide excision repair, an enzyme complex recognizes the distortion in the DNA complex around the thymine dimer and cuts and removes the damaged DNA strand. The correct nucleotides are replaced by DNA pol I and the nucleotide strand is sealed by DNA ligase. (b) In photoreactivation, the enzyme photolyase binds to the thymine dimer and, in the presence of visible light, breaks apart the dimer, restoring the base pairing of the thymines with complementary adenines on the opposite DNA strand. CC BY-SA 4.0 from Parker
SOS-response and Translesion Synthesis
Bacterial cells that have suffered extensive DNA lesions are rescued by the SOS response. There are at least 50 genes that are members of the SOS regulon. Expression of SOS genes is repressed by binding of LexA homodimers to upstream SOS boxes (operator) during normal growth conditions and in the absence of DNA damage (Fig. 6.6). When there is extensive DNA damage, ssDNA forms as a result of DNA polymerase stalling. RecA binds to ssDNA to form RecA nucleoproteins which then associates with and promotes autoproteolytic cleavage of LexA to non-functional subunits and degradation by proteases. The cleaved LexA falls off from the SOS boxes, allowing transcription of the SOS genes. One SOS regulon member codes for UmuDC. UmuC, in a complex with autocleaved UmuD’, forms DNA polymerase V which incorporates incorrect bases in a non-template-based manner. Thus, the newly synthesized DNA contains many lesions; this error-prone repair process is called translesion synthesis.
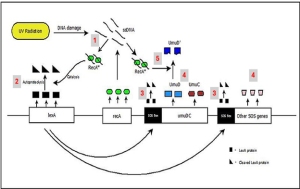
Figure 6.6. SOS-response in response to DNA lesions in E. coli. Extensive DNA damage leads to ssDNA to which RecA binds to form RecA nucleoprotein (1). RecA nucleoprotein associates with LexA dimers ( ▬ ) and (2) promotes autoproteolysis of LexA to non-functional subunits ( ◣). (3) The autocleaved LexA leaves the SOS boxes to allow transcription of SOS genes (4) which include the UmuDC operon. (5) Upon association with RecA nucleoprotein, UmuD gets autocleaved to UmuD’ which then associates with UmuC and other subunits to form an error-prone DNA polymerase complex. CC BY-SA 4.0 from Ragavi Prathyusha
6.4 Identifying Bacterial Mutants
One common technique used to identify bacterial mutants is called replica plating. This technique is used to detect nutritional mutants, called auxotrophs, which have a mutation in a gene encoding an enzyme in the biosynthesis pathway of a specific nutrient, such as an amino acid. As a result, whereas wild-type cells retain the ability to grow normally on a medium lacking the specific nutrient, auxotrophs are unable to grow on such a medium. During replica plating (Fig. 6.7), a population of bacterial cells is exposed to mutagens (e.g. UV light) and then plated as individual cells on a complex nutritionally complete plate, like Luria-Bertani agar (LB agar) and allowed to grow into colonies. Cells from these colonies are removed from this master plate, often using sterile velvet. This velvet, containing cells, is then pressed in the same orientation onto plates of various media. At least one plate should also be nutritionally complete to ensure that cells are being properly transferred between the plates. The other plates lack specific nutrients, like amino acids, thereby allowing the researcher to discover various auxotrophic mutants unable to produce specific nutrients. Cells from the corresponding colony on the nutritionally complete plate can be used to recover the mutant for further study.
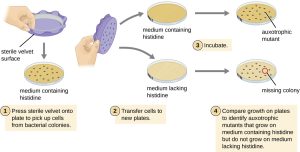
Figure 6.7. Identification of auxotrophic mutants, like histidine auxotrophs, is done using replica plating. After mutagenesis, colonies that grow on nutritionally complete medium but missing on medium lacking histidine are identified as histidine auxotrophs. CC BY-SA 4.0 from Parker
The Ames Test
The Ames test, developed by Bruce Ames (1928–) in the 1970s, is a method that uses bacteria for rapid, inexpensive screening of the carcinogenic or mutagenic potential of new chemical compounds under development, including candidate human drugs. The test measures the rate of reversion mutations associated with exposure to the test compound, which, if elevated, may indicate that exposure to this compound is associated with greater cancer risk. The Ames test uses as the test organism a strain of Salmonella typhimurium that is a histidine auxotroph, that is, unable to synthesize its own histidine because of a mutation in an essential gene required for histidine synthesis. After exposure to a potential mutagen, these bacteria are plated onto a medium lacking histidine, and the number of mutants regaining the ability to synthesize histidine is recorded and compared with the number of such mutants that arise in the control cells which were not exposed to a mutagen (Fig. 6.8). Chemicals that are more mutagenic will bring about more mutants with restored histidine synthesis in the Ames test. Because many chemicals are not directly mutagenic but are metabolized to mutagenic forms by liver enzymes, rat liver extract is commonly included at the start of this experiment to mimic liver metabolism. After the Ames test is conducted, compounds identified as mutagenic are further tested for their potential carcinogenic properties by using other models, including animal models like mice and rats.
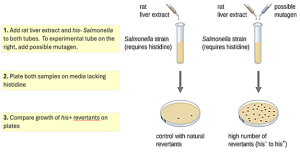
Figure 6.8. The Ames test is used to identify mutagenic, potentially carcinogenic chemicals. A Salmonella histidine auxotroph is used as the test strain, exposed to a potential mutagen/carcinogen. The number of reversion mutants growing in the absence of histidine is counted and compared with the number of natural reversion mutants that arise in the absence of the potential mutagen. More colonies growing on the plate that have bacteria exposed to the test chemical indicate that the chemical is a mutagen. CC BY-SA 4.0 from Parker
References:
1. K.R. Tindall et al. “Changes in DNA Base Sequence Induced by Gamma-Ray Mutagenesis of Lambda Phage and Prophage.” Genetics 118 no. 4 (1988):551–560.
2.Figures and texts in this chapter are mostly adapted or modified from Nina Parker, Mark Schneegurt, Anh-Hue Thi Tu, Philip Lister, Brian M. Forster by CC BY-SA 4.0. Access for free at https://openstax.org/books/microbiology/pages/1-introduction
CHAPTER REVIEW QUESTIONS:

{kind=link}