
8 Chapter 8. Recombinant DNA Technology
Albert B. Flavier
Chapter outline
8.1. Recombinant DNA Technology
8.3. Ligase-independent cloning
Learning Objectives
By the end of this chapter, you would be able to:
-
Describe the different enzymes used in making recombinant DNA
-
Describe strategies to increase efficiency of recombinant DNA construction
-
Describe different types of DNA libraries and their construction
- Describe ligase-independent methods of constructing recombinant DNA
8.1 RECOMBINANT DNA TECHNOLOGY
Biotechnology is the use of technology to modify or improve organisms or their products for the benefit of man. Modern approaches to modifying organisms commonly involve genetic engineering, the alteration of an organism’s genetic makeup. Genetic engineering involves the use of recombinant DNA technology (RDT), the process by which DNA fragments from two different sources are combined (Fig. 8.1). The recombinant DNA is usually constructed in vitro before introduction into an organism. If the DNA that is introduced comes from a different species, the host organism is now considered to be transgenic. In the genetic engineering of many organisms, the recombinant DNA is first transformed and propagated in Escherichia coli and confirmed to have the correct sequence before introduction into the final organisms to be genetically engineered.
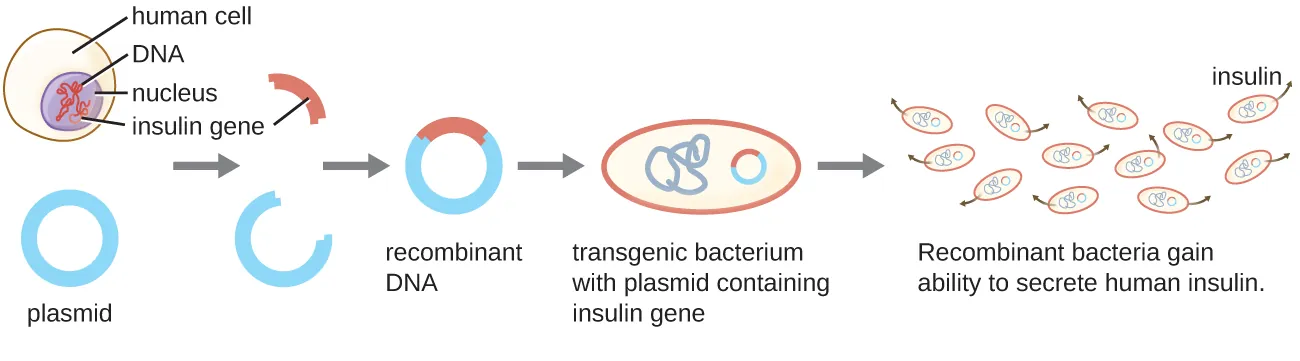
Figure 8.1. Recombinant DNA technology is the artificial joining together of DNA fragments from two sources. In the above example, the human insulin gene is inserted into a bacterial plasmid. The recombinant plasmid is then transformed into bacteria, which gain the ability to produce many copies of the insulin protein. CC-SA-4.0 from Parker
Recombinant DNA technology is often interchanged with the use of the term molecular cloning. Molecular cloning is the production of many copies of biomolecules such as DNA, RNA, or protein, often by employing RDT. Polymerase chain reaction (PCR) is one molecular cloning approach that makes billions of copies from a single molecule of a target DNA fragment. The propagation of a recombinant plasmid carrying the gene/s of interest in a bacterial host and/or the resulting production of the target protein/s are other examples of molecular cloning. Herbert Boyer and Stanley Cohen first demonstrated the complete molecular cloning process in 1973 when they successfully cloned genes from the African clawed frog (Xenopus laevis) into a bacterial plasmid that was then introduced into the bacterial host, Escherichia coli.
Restriction Enzymes and Ligases
The creation of recombinant DNA molecules is possible due to the use of naturally occurring restriction enzymes (restriction endonucleases), bacterial enzymes produced as a protective mechanism to cut and destroy foreign DNA that is introduced during bacteriophage infection. Stewart Linn and Werner Arber discovered restriction enzymes in their 1960s studies of how E. coli limits bacteriophage replication upon infection. Today, we use restriction enzymes extensively for cutting DNA fragments that can then be spliced into another DNA molecule to form recombinant molecules. Each restriction enzyme cuts DNA at a characteristic recognition site, a specific, usually palindromic, DNA sequence typically between four to six base pairs in length. A palindrome is a sequence of letters that reads the same forward as backward. (The word “level” is an example of a palindrome.) Palindromic DNA sequences contain the same base sequences in the 5ʹ to 3ʹ direction on one strand as in the 5ʹ to 3ʹ direction on the complementary strand. Many restriction enzymes work as homodimers (Fig. 8.2), where one subunit recognizes and cuts the DNA palindrome in one strand, and the other subunit cuts the opposite strand at an identical position in the palindrome. Some restriction enzymes, like EcoRI, cut to produce molecules that have complementary overhangs (sticky ends) while others, like SmaI, cut without generating such overhangs, producing blunt ends instead (Fig. 8.2). Restriction enzymes that recognize 4 base pairs are monomers, while those that recognize 8 base pairs or more are heterotetramers.

Figure 8.2. In the first six-nucleotide restriction enzyme site recognized by the enzyme, EcoRI, notice that the sequence reads the same in the 5ʹ to 3ʹ direction on both strands. This is known as a palindrome. The cutting of the DNA by EcoRI at the sites indicated by the green line produces DNA fragments with sticky ends. The second six-nucleotide recognition site also exhibits a palindromic sequence. The cutting of the DNA by the restriction enzyme SmaI at the green line produces DNA fragments with blunt ends. Any other piece of blunt DNA could attach to one of the blunt ends produced, forming a recombinant DNA molecule. Adapted from www.neb.com (2020 catalog) with permission from New England Biolabs.
Molecules with complementary sticky ends can easily anneal, or form hydrogen bonds between complementary bases in the opposite DNA strands. The joining together of two single strands of DNA with complementary sequences is referred to as hybridization. Blunt ends can also attach together, but less efficiently than sticky ends due to the lack of complementary overhangs to facilitate the process. In either case, ligation by DNA ligase can then rejoin the two sugar-phosphate backbones of the DNA through covalent bonding, making the molecule a continuous double strand. In 1972, Paul Berg was the first to produce a recombinant DNA molecule using this technique, combining the SV40 monkey virus with E. coli bacteriophage.
Plasmids
After restriction digestion, genes of interest are commonly inserted into plasmids, small pieces of typically circular, double-stranded DNA that replicate independently of the bacterial chromosome. In recombinant DNA technology, plasmids are often used as DNA vectors to carry DNA fragments from one organism to another. Plasmids used as vectors can be genetically engineered by researchers and scientific supply companies to have specialized properties, as illustrated by the commonly used plasmid vector pUC19 (Fig. 8.3). Many plasmid vectors contain genes that confer antibiotic resistance; these resistance genes facilitate selection of plasmid-containing colonies by plating them on media containing the corresponding antibiotic. The antibiotic kills all cells that do not harbor the desired plasmid vector, but those that contain the vector are able to survive and grow.
Plasmid vectors used for cloning typically have a polylinker site, or multiple cloning site (MCS) (Fig. 8.3). A polylinker site is a short sequence containing multiple unique restriction enzyme recognition sites that are used for inserting another DNA fragment into the plasmid after restriction digestion of both the DNA fragment and the plasmid. Having multiple restriction enzyme recognition sites within the polylinker allows different cloning experiments involving different restriction enzymes.
This polylinker site is often found within a reporter gene, another gene sequence artificially engineered into the plasmid that encodes a protein that allows for visualization of successful DNA insertion. The reporter gene allows a researcher to distinguish (screen) host cells that contain recombinant plasmids with cloned DNA fragments from host cells that only contain the non-recombinant plasmid vector. The most common reporter gene used in plasmid vectors is the bacterial lacZ gene encoding beta-galactosidase, an enzyme that naturally degrades lactose but can also degrade a colorless synthetic analog X-gal, thereby producing blue colonies on X-gal–containing media. The lacZ reporter gene gets disabled when another DNA fragment is inserted into the plasmid. Because the LacZ protein is not produced when the gene is disabled, X-gal is not degraded and white colonies are produced, which can then be isolated.
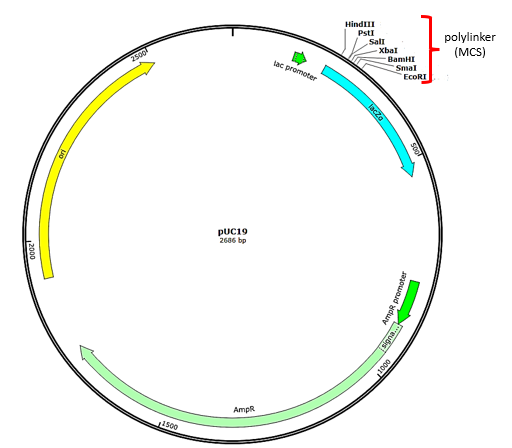
Figure 8.3. Features found in the plasmid vector pUC19. Arrows indicate the directions in which the genes are transcribed. The polylinker contains multiple unique restriction enzyme recognition sites and is found within the lacZ reporter gene (turquoise). Also note the ampicillin resistance gene (AmpR – light green) encoded on the plasmid. Plasmid map generated using SnapGene.
DNA Transformation
The most commonly used mechanism for introducing engineered plasmids into a bacterial cell is by transformation (Fig. 8.4), a process in which bacteria take up free DNA from their surroundings. Some bacteria, such as Bacillus spp., are naturally competent in taking up foreign DNA. In most cases, bacteria must be made artificially competent in the laboratory by increasing the permeability of the cell membrane. This can be achieved through chemical treatments that neutralize charges on the cell membrane or by exposing the bacteria to an electric field that creates microscopic pores in the cell membrane. These methods yield chemically competent or electrocompetent bacteria, respectively.
Following the transformation protocol, bacterial cells are plated onto medium containing an antibiotic to inhibit the growth of host cells that were not transformed by the plasmid that would have conferred antibiotic resistance. A technique called blue-white screening (Fig. 8.5) is then used for lacZ-encoding plasmid vectors such as pUC19. Blue colonies have a functional beta-galactosidase enzyme because the lacZ gene is uninterrupted when no foreign DNA got inserted into the polylinker site. These colonies typically result from the digested, linearized plasmid religating onto itself. White colonies lack a functional beta-galactosidase enzyme which happens when foreign DNA gets successfully inserted within the polylinker site of the plasmid vector, thus disrupting the lacZ gene. The recombinant plasmids can be isolated from the white colonies correct and the DNA insert sequenced for confirmation.
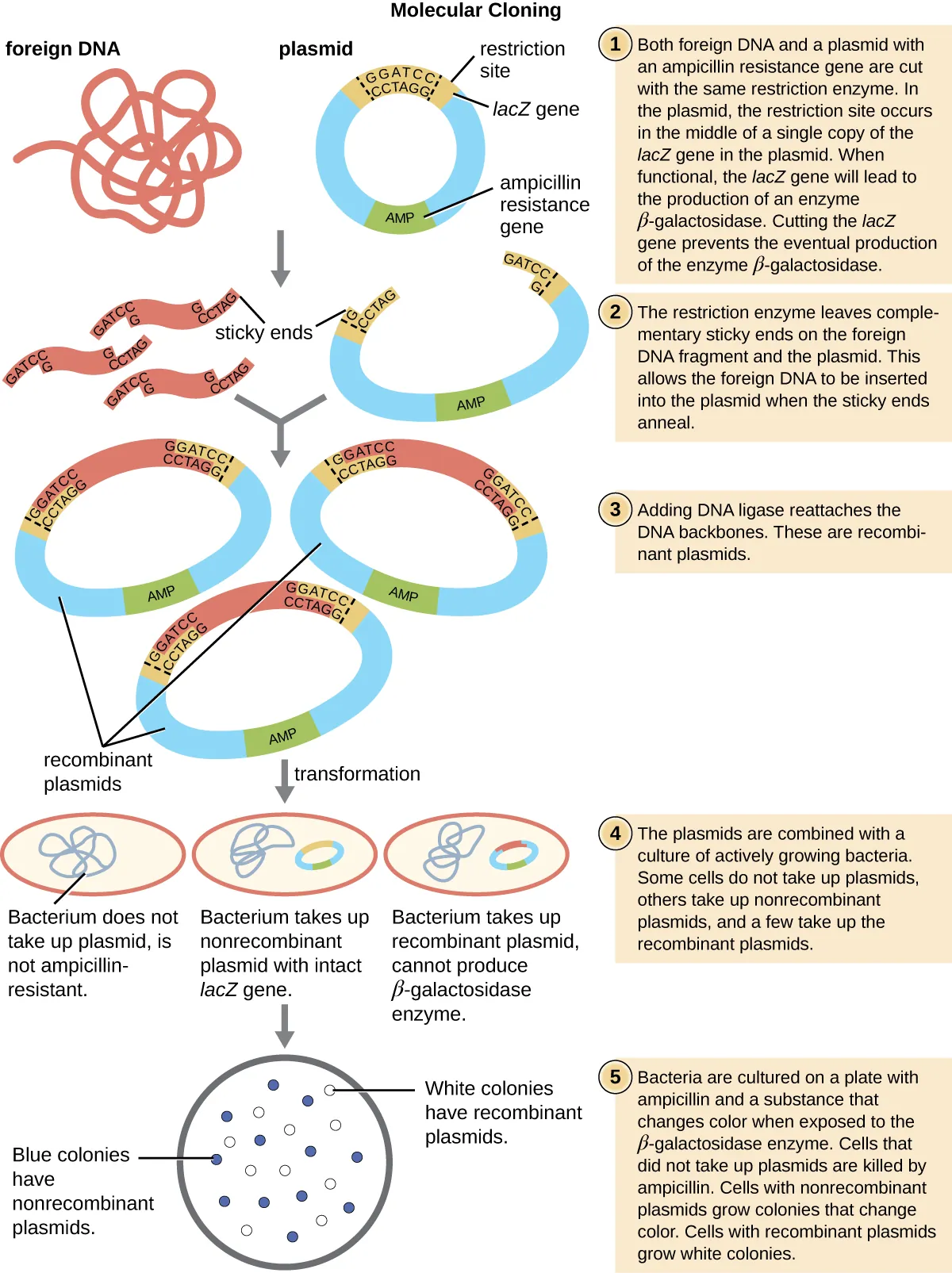
Figure 8.4 The steps involved in molecular cloning using bacterial transformation, followed by blue-white screening, are outlined in this graphic flowchart. From Parker
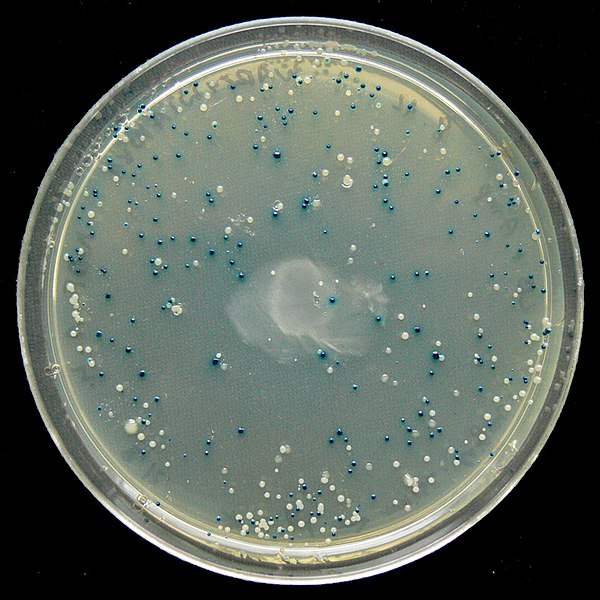
Figure 8.5. Blue-white screening of E. coli transformed with pUC19 and plated on media with ampicillin and the b-galactosidase substrate, X-Gal. Blue colonies carry non-recombinant pUC19 with no inserted DNA fragment interrupting the lacZ gene, in which case the functional b-galactosidase hydrolyzes X-Gal from colorless to an insoluble blue chemical. White colonies carry pUC19 with a DNA fragment inserted within lacZ thus interrupting production of functional b-galactosidase. CC-SA-BY-4.0 Walkowski
Increasing ligation efficiency
The presence of many blue colonies in transformation plates shows that ligation to produce recombinant DNA molecules is not 100% efficient. There are several strategies to improve the efficiency of recombinant DNA construction.
- Increase the ratio of insert to plasmid
- Dephosphorylation of the cloning vector
- Two-enzyme digest
Increasing the ratio of DNA fragment to plasmid
In a ligation reaction mixture, where the plasmid and DNA fragment were digested with one restriction enzyme, intramolecular ligation or self-ligation of the plasmid is preferred because two ends of the cut plasmid are in one molecule have higher odds of finding each other. To increase intermolecular ligation between plasmid and gene of interest, an optimal ratio of 3:1 molar ratio of insert:plasmid is often suggested. Ratios from 2:1 to 10:1 can be tested also.
Dephosphorylation of the cloning vector
A second approach to decrease self-ligation of a plasmid that was cut with a single restriction enzyme is to dephosphorylate the digested cloning vector (Fig. 8.6a) by using alkaline phosphatase. Cutting the plasmid with restriction enzymes results to breaking of the phosphodiester bond in the two strands of DNA leaving a phosphate group in the 5’ end and a hydroxyl group in the 3’ end (Fig. 8.6) of both DNA strands. DNA ligase catalyzes the condensation reaction that leads to the reformation of the broken phosphodiester bonds. Dephosphorylating the cut plasmid removes the 5’ phosphate groups from both DNA strands such that reformation of phosphodiester bonds between the 3’ hydroxyl and 5’ dephosphorylated ends are no longer possible. The incoming DNA fragment (GOI or gene of interest) have 5’ phosphate groups, that can form phosphodiester bonds with the hydroxyl groups in the dephosphorylated plasmid (Fig 8.6b). Because the phosphate groups are missing from the plasmid ends, no phosphodiester bonds can form between the plasmid 5’ ends and the GOI 3’ hydroxyl ends, there are nicks that will remain in both DNA strands. How is the phosphodiester bond restored in the remaining nicks? Once the nicked recombinant plasmids are transformed into E. coli, a cytoplasmic enzyme will catalyze the formation of the phosphodiester bonds in the nicks.


Figure 8.6. Dephosphorylation of cloning vector using alkaline phosphatase. The 3’ hydroxyl groups are represented by blue dots; the 5’ phosphates represented by red dots. 1. Religation of restriction enzyme digested plasmid in the presence of DNA ligase. Black arrows are phosphodiester bonds formed upon religation of vector ends. 2. Alkaline phosphatase treatment of cut plasmid removes the 5’ phosphates. In the presence of ligation, Xs are nicked strands where no phosphodiester bonds are restored and therefore no religation of the plasmid ends takes place. 3. Mixing of alkaline phosphatase treated plasmid with DNA fragment containing the gene of interest in the presence of DNA ligase leads to successful ligation of the gene of interest. Open triangles are nicks where no phosphodiester bonds are formed in vitro, but are repaired upon transformation into E. coli. Flavier (CC0 1.0).
Two-enzyme digest to increase recombinants
The third approach that will favor formation of recombinant plasmids is to use two different enzymes to generate incompatible ends in the vector. Dephosphorylation may be omitted when using this two-enzyme approach. Figure 8.3 in pUC19 shows an example of a multiple cloning site (MCS) that is found in cloning vectors. MCS in plasmids contains multiple restriction enzyme sites many of which are not found anywhere else in the plasmid. One can choose from the restriction sites where to insert the GOI. In pUC19, the plasmid can be cut with EcoRI to generate a sticky overhang, and SmaI to generate a blunt end, resulting in plasmid ends that are not compatible (Fig. 8.7), and therefore won’t get religated. If the GOI is cut with the same two enzymes, EcoRI and SmaI, the insert now has one overhang that is compatible with the EcoRI overhang, while the blunt end of the fragment is compatible with the SmaI-generated blunt end in the plasmid. The blunt cutter enzyme can be any blunt cutter and does not have to be SmaI; the blunt ends would always be compatible.

Figure 8.7. Using two different restriction enzymes to minimize plasmid religation. The two enzymes used should produce incompatible ends. The top image shows the nucleotides recognized by one restriction enzyme, EcoRI, that produces a sticky overhang and a second enzyme, SmaI, which generates blunt ends; red triangles are the restriction enzyme cut sites. The ends are not compatible and the plasmid could not religate onto itself. In the lower figure, a DNA fragment cut with the same two enzymes have ends compatible with ends of the cut plasmid and should get ligated successfully in the plasmid. Flavier (CC0 1.0).
Directional cloning using two enzymes
When cutting with a single restriction enzyme, because both ends of the GOI fragment and the plasmid ends will contain the same overhangs, the GOI can get inserted in two different orientations with respect to the promoter in the plasmid. Around fifty percent of the time, the GOI will get oriented backwards, so that its 3’ end can will be closer to the promoter region. In that case, the gene of interest will not be transcribed and translated properly. Using two restriction enzymes will facilitate directional cloning in the vector with respect to the direction of the promoter and translation control regions. In the pUC19 MCS (Fig.8.3), the SmaI site is closer to and downstream from the lac promoter, while the EcoRI site is further downstream. Therefore, the 5’ end of the GOI should contain the SmaI site and the EcoRI site situated in the 3’ end of the GOI. Another pair of enzymes that can be used for directional cloning are PstI and BamHI, with the PstI site in the 5’ end of the GOI and BamHI in the 3’ end. Directional cloning can be used also to add extra amino acid sequences to the GOI such as purification tags (HIS-tags) or translational reporters (GFP).
Adding restriction enzyme cut sites by PCR
When a DNA fragment needs to be properly oriented with respect to the promoter in the plasmid, two different restriction enzyme cut sites can be introduced to the DNA fragment during PCR (Fig. 8.8). The restriction sites are added to the PCR products by incorporating the cut sites in the 5’ ends of both forward and reverse PCR primers. Once amplified by PCR, the resulting PCR products will have two different restriction cut sites incorporated in the fragment ends. Note that PCR primers don’t have to be 100% complementary throughout their entire length to the target sequence; 20-35 nucleotides in the primer 3’ ends complementary to the target sites are sufficient. Mismatches in the 5’ ends are tolerated in the PCR reaction.
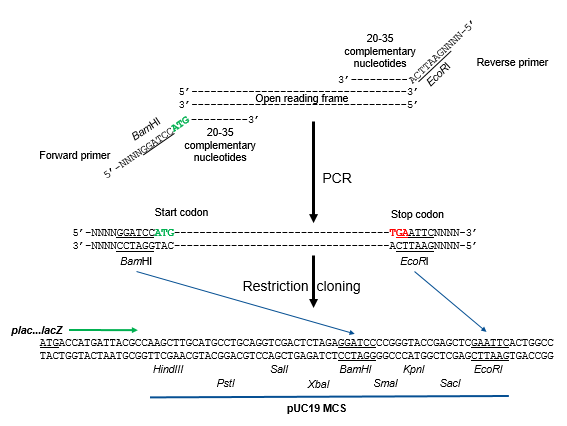
Figure 8.8. Directional cloning and PCR. The open reading frame (or gene of interest) is amplified by PCR using forward and reverse primers where the sequences in their 5’ ends will introduce two different restriction enzyme cut sites (BamHI and EcoRI) for directional cloning in the MCS of a plasmid (pUC19 in this example). Sequences representing start (green) and stop codons (red) are also added in the 5’ portions of the primers. The 3’ ends of the primers are complementary to the open reading frame to be amplified. The PCR products are either digested simultaneously or sequentially with BamHI and EcoRI, then ligated into similarly digested pUC19. Upon ligation, the start codon is positioned to be closer to, and the same direction as, the lac promoter, plac, for successful transcription and translation. Flavier (CC0 1.0).
8.2 DNA Libraries
Molecular cloning may be used to generate a genomic library. A genomic library is a complete (or nearly complete) collection of fragments of an organism’s genome contained as recombinant DNA plasmids engineered into unique clones of bacteria. Having such a library allows a researcher to create large quantities of each fragment by growing the bacteria carrying that fragment. These fragments can be used to determine the sequence of the inserted DNA and the function of any genes present.
One method for generating a genomic library is to ligate individual restriction enzyme-digested genomic fragments into plasmid vectors cut with the same restriction enzyme (Fig. 8.9). During transformation, each transformed bacterial cell takes up a single recombinant plasmid and grows into a colony of cells. All of the cells in each colony are identical clones and carry the same recombinant plasmid (same insert). The resulting library is a collection of colonies, each of which contains a fragment of the original organism’s genome, that are each separate and distinct and can each be used for further study. This makes it possible for researchers to screen these different clones to discover the one containing a gene of interest from the original organism’s genome.
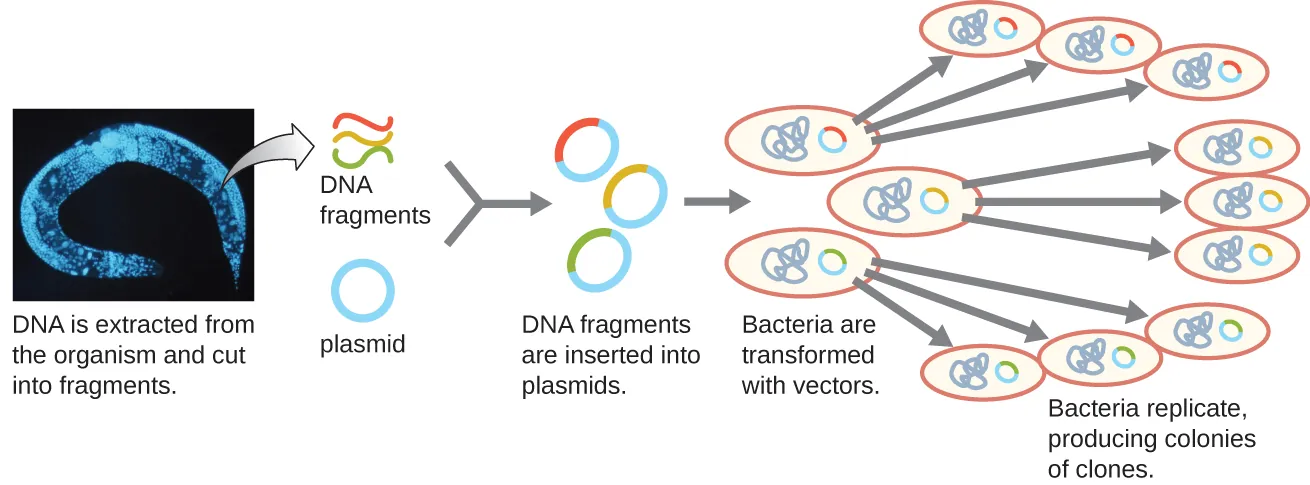
Figure 8.9. Genomic library construction. The generation of a genomic library facilitates the discovery of the genomic DNA fragment that contains a gene of interest. (credit “micrograph”: modification of work by National Institutes of Health). CC-SA-4.0 from Parker
To construct a genomic library using larger fragments of genomic DNA, an E. coli bacteriophage, such as lambda, can be used as a host (Fig. 8.10). Genomic DNA can be sheared or enzymatically digested and ligated into a pre-digested bacteriophage lambda DNA vector. The resulting recombinant phage DNA molecules can be packaged into phage particles and used to infect a dense lawn of E. coli on a plate. During infection within each cell, each recombinant phage will make many copies of itself and lyse the E. coli lawn, forming a plaque. Thus, each plaque from a phage library represents a unique recombinant phage containing a distinct genomic DNA fragment. Plaques can then be screened further to look for genes of interest. One advantage to producing a library using phages instead of plasmids is that a phage particle holds a much larger insert of foreign DNA compared with a plasmid vector, thus requiring a much smaller number of cultures to fully represent the entire genome of the original organism.
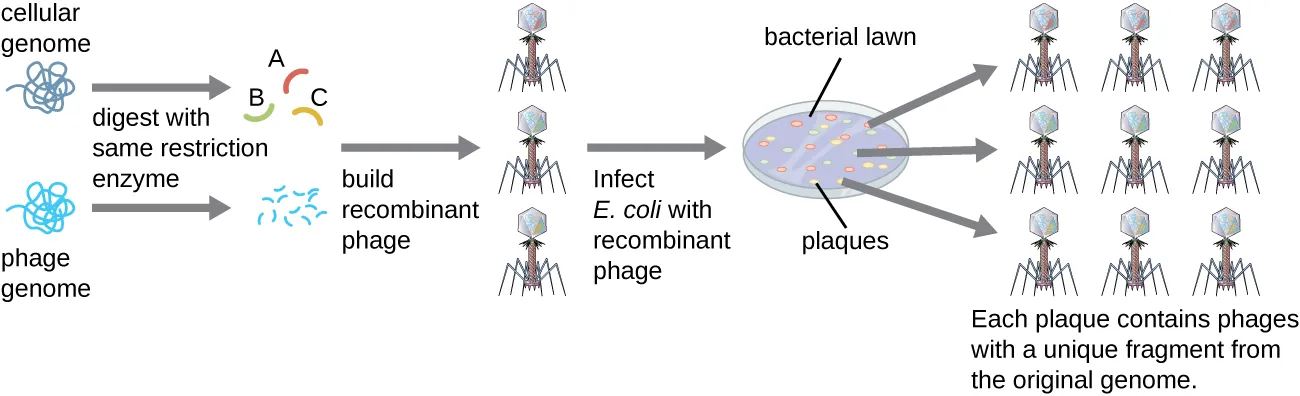
Figure 8.10. Phage libraries. Recombinant phage DNA molecules are made by ligating restriction-enzyme digested phage DNA with fragmented genomic DNA molecules. These recombinant phage DNA molecules are packaged into phage particles in vitro and allowed to infect a bacterial lawn. Each plaque represents a unique recombinant DNA molecule that can be further screened for genes of interest. CC-SA-4.0 from Parker
To focus on the set of differentially expressed genes in an organism or even a tissue, researchers construct libraries using the organism’s messenger RNA (mRNA) rather than its genomic DNA. Whereas all cells in a single organism will have the same genomic DNA, different tissues express different genes, producing different complements of mRNA. For example, all human cells’ genomic DNA contains the gene for insulin, but only cells in the pancreas express mRNA directing the production of insulin. Because mRNA cannot be cloned directly, in the laboratory mRNA must be used as a template by the retroviral enzyme reverse transcriptase to make complementary DNA (cDNA). A cell’s full complement of mRNA can be reverse-transcribed into cDNA molecules, which can be used as a template for DNA polymerase to make double-stranded DNA copies; these fragments can subsequently be ligated into either plasmid vectors or bacteriophage to produce a cDNA library. The benefit of a cDNA library is that it contains DNA only from the expressed genes in the cell. This means that the introns, control sequences such as promoters, and DNA not destined to be translated into proteins are not represented in the library. The focus on transcribed sequences means that the library cannot be used to study the sequence and structure of the genome in its entirety. The construction of a cDNA genomic library is shown in Figure 8.11.
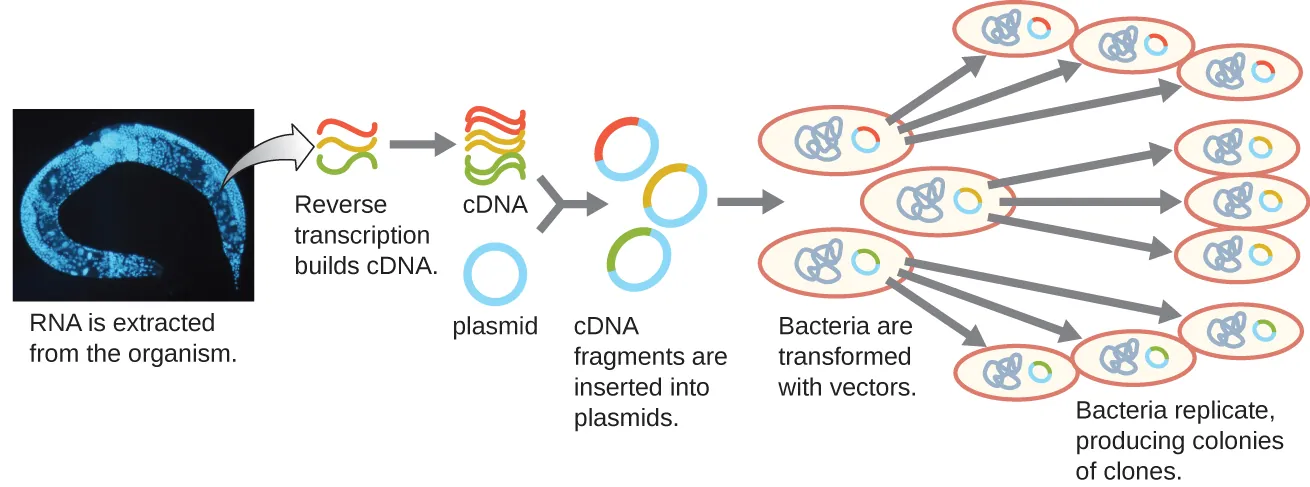
Figure 8.11. cDNA library construction. mRNA is extracted from an organism. Complementary DNA (cDNA) is made from mRNA by the retroviral enzyme reverse transcriptase, converted into double-stranded copies, and ligated into plasmid vectors. (credit “micrograph”: modification of work by National Institutes of Health). CC-SA-4.0 from Parker
Colony blots
In genomic and cDNA libraries, there are millions of colonies with different DNA fragments cloned. To find a specific gene one is interested in, a common technique used is colony blotting or hybridization (Fig. 8.12). Say in yeast there is a gene required for cell division and you want to find a possible homologue in another organism like an ostrich. To find a yeast homologue from an ostrich genomic or cDNA library, E. coli transformants are plated on selective media. A membrane (nitrocellulose or nylon) is laid on top of the E. coli colonies, whereupon the sticky cells will get transferred to the membrane. The bacterial cells stuck on the membrane are then lysed using sodium hydroxide and SDS, thus releasing the denatured single-stranded DNA. The released DNA is crosslinked to the membrane by exposure to UV light. A single-stranded nucleic acid probe, whose sequence is completely or partially complementary to the target gene, is added to buffer and allowed to hybridize to single-stranded DNA on the membrane. The probe can be fluorescently labeled, radiolabeled with 32P, or has an enzyme conjugated. If there’s a yeast homologue in the library, the DNA probe will hybridize with spots on the membrane that correspond with the colonies on the plate that carry the GOI. For radio- or fluorescently labeled probes, the hybridizing spots can be visualized by autoradiography (by exposing an X-ray film) or using an equipment called phosphorimager (Watch the video below). Enzyme-labeled probes are incubated with a chemiluminescent substrate prior to exposing an X-ray film to the blot.
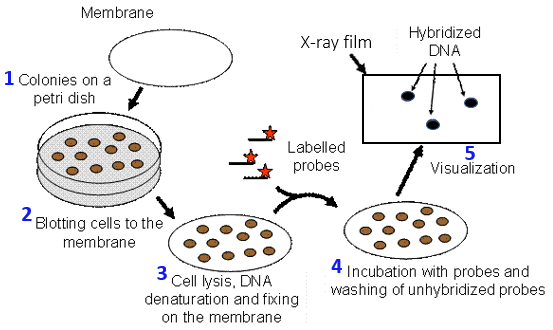
Figure 8.12 Colony blotting to screen a DNA library. 1) E. coli cells containing the DNA library are plated on selective media. 2) A membrane (nitrocellulose or nylon) is laid on top of the colonies, and cells get transferred to the membrane. 3) Cells on the membrane are lysed by soaking the membrane in a solution containing sodium hydroxide and SDS, thus releasing the denatured single-stranded DNA. The DNA is crosslinked to the membrane by exposure to UV light. 4) A labelled probe whose sequence is complementary to the target gene, is allowed to hybridize to the DNA on the membrane. 5) The hybridizing spots can be visualized by exposing an X-ray film to the membrane. Image released to public domain by Kaksonen
Watch: Using a phosphorimager:
The hybridizing colony possibly carrying the gene of interest is inoculated into liquid media with antibiotic and the plasmid purified. The library insert can be sequenced to pinpoint the GOI, or a Southern blot of a restriction enzyme digest of the plasmid can be used to identify the GOI which can be subcloned. GOI-specific primers can be used also to PCR out the yeast gene homolog for subsequent cloning into an expression vector for protein production.
8.3. Ligase-independent cloning methods
In traditional recombinant DNA construction, DNA ligase is used to join together a DNA fragment with a plasmid. Newer techniques do not make use of DNA ligase, and some do away with the use of restriction enzymes. These techniques can be easier and more efficient than traditional techniques. The techniques are particularly useful when there are no convenient restriction sites available in the GOI or in the cloning vector. In certain cases, a cloning vector may only accept inserts with blunt ends. However, cloning of blunt-ended fragments is a very inefficient process leading to many self-ligated plasmids without inserts. Some widely used ligase-independent techniques are:
1. TA cloning
2. Gateway cloning
3. Recombineering
4. Exonuclease-mediated cloning
1. TA-cloning
TA-cloning uses a plasmid prepared to have single thymine (T) overhangs on the 3’ ends of both DNA strands. In one version of TA cloning, i.e., Topo cloning, a topoisomerase is conjugated to the phosphate group of both T overhangs. The DNA fragment to be inserted needs to have 5’ adenine (A) overhangs which are complementary to the T on the plasmid ends. The A overhangs are usually generated by PCR using Taq polymerase since Taq polymerase preferentially generates PCR products with 5’ A overhangs; other thermostable enzymes do not produce the A overhangs. When the plasmid and the PCR products are mixed, the complementary T and A overhangs hybridize and the topoisomerase catalyzes the formation of phosphodiester bonds between the two molecules. One disadvantage of Topo cloning is that the preparation of the topoisomerase-activated vector is time consuming. There are commercially available topoisomerase-activated vectors, but they are of limited types, and can be pricey compared to standard home-made plasmids. In another version of TA cloning, a topoisomerase is not conjugated to the T. Instead, DNA ligase is added to the reaction mixture and catalyzes formation of phosphodiester bonds between the A and T in the plasmid and the insert.
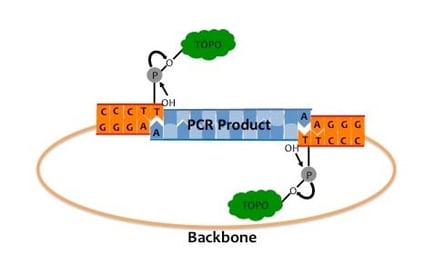
Figure 8.13. TOPO TA cloning of Taq-amplified DNA. Topoisomerase (TOPO) is covalently bound to the 3’ phosphates in the T overhangs in both ends of the linearized vector. In the presence of Taq-amplified PCR product with A overhangs, topoisomerase catalyzes ligation of the plasmid and PCR product ends. Adapted from Swanson.
2. Gateway cloning
Gateway cloning (Fig. 8.14) makes use of site-specific recombination between attP sequences, originally found in lambda bacteriophage, and attB, the site of lambda phage DNA integration in the E. coli chromosome. The GOI is PCR-amplified with primers containing attB sequences incorporated on their 5’ ends, then the PCR product is recombined into a donor vector containing a ccdB gene flanked by attP sequences. ccdB codes for a toxic protein, CcdB, that poisons DNA topoisomerase II to interfere with DNA replication. When incubated with a BP Clonase, the GOI attBs recombine with the plasmid attPs, with the GOI replacing ccdB. Thus, only transformants that carry the vector:GOI recombinants will grow in selective media. The donor vector, now carrying the GOI, is then referred to as entry vector, because from there, the GOI can be recombined into other Gateway “destination vectors” that contain many different vector features, e.g., different selection markers, promoters, fusion tags for purification, etc. Recombination between entry and destination vectors occur in the presence of LR Clonase. There are different Gateway destination vectors for protein expression in different cell systems other than E. coli – yeast, insect or mammalian cells.
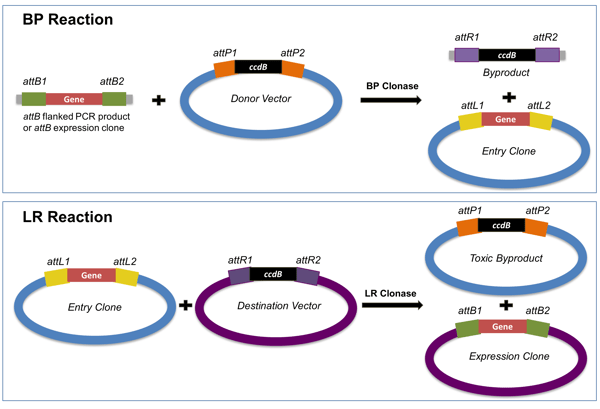
Figure 8.14. The Gateway system adopts phage integration into the BP and LR reactions. The BP reaction recombines the gene of interest into the donor vector and creates an entry clone where the GOI is flanked with attL (upper image). The LR reaction inserts the GOI into the destination vector and creates an expression clone with all of the components necessary for expression of the gene into protein in a target organism. Adapted from Soriano.
3. Exonuclease-mediated cloning
In one example of exonuclease-mediated cloning (Fig. 8.15), ligation-independent (LIC) vectors with 5’ overhangs are prepared by treatment of a restriction-enzyme linearized plasmid with T4 (or Pfu) DNA polymerase in the presence of excess dTTP. The polymerase has 3’ to 5’ exonuclease activity. The excess dTTP will favor incorporation of dTTP over the removal of T from the DNA ends and so will stop exonuclease activity, thus leaving 5’ overhangs. Sequences identical and complementary to the plasmid overhangs are incorporated into the 5’ ends of the forward and reverse PCR primers, while the 3’ ends of the primers are specific for annealing to GOI sequences. The PCR product which has the GOI is treated with Pfu DNA polymerase, but this time, dATP in the reaction buffer stops exonuclease activity of DNA polymerase when an A is reached as the excess dATP will favor incorporation of dATP over its removal from the ends. Once mixed, the GOI fragment and vector sticky ends form H-bonds between the complementary bases. Nicks remain between the GOI and vector ends, and the final phosphodiester bonds are formed once the DNA is transformed and inside E. coli.
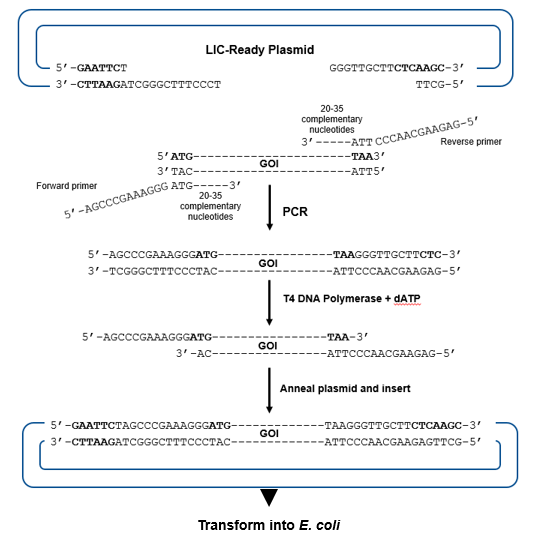
Figure 8.15. Exonuclease-mediated, ligation-independent cloning (LIC). The LIC ready plasmid has 3’ overhangs prepared by digesting with T4 DNA polymerase in the presence of excess dTTP. The insert is prepared by PCR with primers whose 5’ ends would be complementary to the plasmid overhangs and the 3’ ends complementary to the GOI sequences. 5’ overhangs are produced from the PCR product by the 3’ to 5’ exonuclease activity of T4 DNA polymerase in the presence of excess dATP. The plasmid and GOI overhangs are allowed to anneal and transformed into E. coli where phosphodiester bonds form in the nicked strands. Flavier (CC0 1.0).
4. Recombineering using Lambda Red
While Gateway cloning involves site-specific recombination between att sequences, recombineering (recombination-mediated genetic engineering) requires homologous sequences in the GOI fragment and in the plasmid (Fig. 8.16). The homologous sequences in a double-stranded GOI fragment are introduced by PCR where the 5’ ends of both forward and reverse primers contain sequences (`50 nt) homologous to that found in the plasmid, while the 3’ sequences (~20 nt) are complementary to sequences in the GOI to be inserted. The PCR product containing the GOI is electroporated into E. coli carrying the cloning vector and expressing the lambda red bacteriophage genes, exo, beta, and gam. Exo has 5’ to 3’ exonuclease activity that will generate single stranded (ssDNA) 3’ overhangs to which Beta will bind to protect from nuclease attack. Beta-coated DNA (nucleoprotein) will then invade homologous sequences in the plasmid. Gam blocks exonuclease activity of endogenous RecBCD to prevent it from attacking the PCR product. When recombination between the GOI and plasmid occurs, a lethal gene in the plasmid will get replaced with the GOI. Thus, only transformants carrying “recombineered plasmids” will grow on selection plates. Common lethal genes employed include sacB (lethal when sucrose is added to the media), ccdB, and eco471R ( a lethal restriction enzyme). Synthetic, single-stranded GOI fragment can also be recombined and integration will occur in the presence of Beta alone; Exo and Gam are not needed.
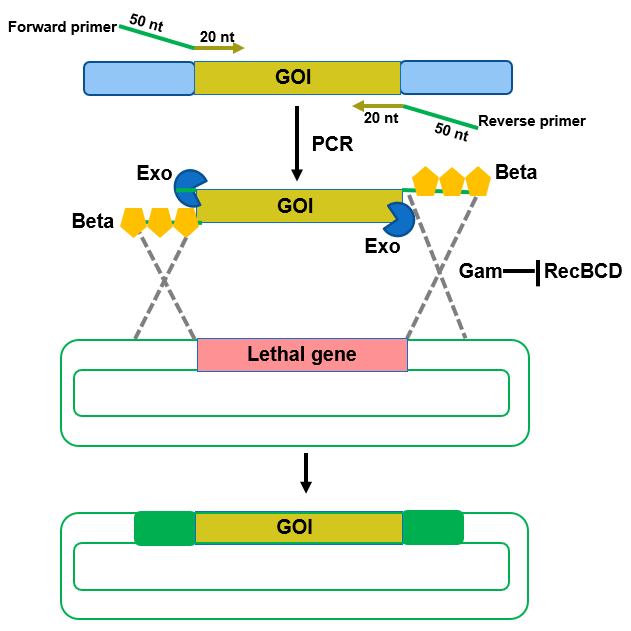
Figure 8.16. Recombineering for gene construction. The GOI which can be amplified by using PCR, recombines with sequences in the plasmid to replace a lethal gene and give rise to the recombinant plasmid with the GOI inserted by using Exo, Beta and Gam from lambda bacteriophage. Modified from Kenkel
Attribution:
Many figures and texts in this chapter are adapted and or modified from OpenStax Parker (CC BY-SA 4.0)
https://openstax.org/books/microbiology/pages/12-1-microbes-and-the-tools-of-genetic-engineering
End-of-Chapter Questions:

{kind=link}
{kind=link}