
9 Chapter 9. Strain Engineering for Recombinant Protein Expression
Hemen Hosseinzadeh and Venkatesh Balan
Chapter Outline
9.2 Expression Systems for Recombinant Proteins
9.3 Strategies for Enhancing Protein Expression
9.4 Metabolic Engineering for Enhanced Protein Production
9.5 Future Perspectives and Emerging Research Areas
Learning Objectives
-
Understand the role and applications of recombinant proteins in medicine, industry, and research.
-
Compare major expression systems (bacteria, yeast, fungi, mammalian, cell-free, novel hosts) and their pros/cons.
-
Apply strategies such as chaperone co-expression, codon optimization, mutagenesis, and engineered strains to enhance protein expression.
-
Use metabolic engineering, CRISPR/Cas9, and synthetic biology tools to optimize host pathways for higher yields.
-
Explore emerging hosts, AI/ML tools, and sustainable production strategies for future protein manufacturing.
9.1 Introduction
Importance of recombinant protein expression in biotechnology and pharmaceuticals
Imagine a world in which we could not produce insulin for diabetics, clotting factors for hemophilia patients, or enzymes that convert plant waste into biofuels. Such a world would lack recombinant protein expression, a cornerstone of modern biotechnology that enables life-saving therapies, sustainable fuels, and modern industrial applications. By inserting a specific gene into a host organism, such as E. coli, yeast, or mammalian cells, we can make it produce valuable proteins that save lives, power industries, or help scientists unlock the secrets of biology. These recombinant proteins are everywhere: in insulin injections, antibodies against cancer, and enzymes that make detergents or biofuels. They have changed medicine by making therapeutic proteins more accessible and revolutionized the industry with tailor-made enzymes for various processes and applications. In research, recombinant proteins serve as indispensable tools for the study of biological mechanisms and the development of drugs. The versatility of this technology lies in its ability to produce proteins from simple hormones to complex antibodies with high precision and scalability. However, success depends heavily on the choice of host organism, which influences protein yield, functionality, and production costs. Figure 9.1 illustrates the core concept of recombinant protein expression and the role of different host cells in meeting specific protein production requirements. So, how do you choose the right host for the job? It’s a bit like choosing the perfect tool for a DIY project: you need to match the tool to the task at hand. Let’s take a look at the most important factors that play a role in this decision.
Criteria for Selecting the Right Host System
Choosing a host system for recombinant protein production is all about finding the sweet spot between biology, practicality, and cost. Here’s what you need to think about:
- What’s the protein like? Proteins vary in complexity. Simple ones like insulin are short chains of amino acids, while others, such as antibodies, require intricate PTMs like glycosylation or disulfide bond formation. If your protein needs these modifications, a eukaryotic host like yeast or mammalian cells is necessary, as they possess the cellular machinery for such processing.
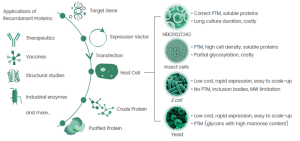
- How much protein do you need? For industrial applications— like enzymes used in detergents or food processing— high yields at low cost are crucial. Bacteria like E. coli are ideal in this case, capable of producing grams of protein per liter of culture. For biopharmaceuticals, however, quantity may be less important than quality, prompting the use of more specialized hosts that ensure correct folding and functionality.
- What is your budget? Cost matters. Culturing E. coli is inexpensive, often costing just pennies per liter. In contrast, mammalian cell cultures can range from $5 to $50 per liter due to their complex media and slower growth. If budget constraints are a concern, bacteria or yeast are economical choices. For high-value therapeutics, though, the added expense of mammalian systems may be worthwhile.
- Is it safe for humans? Safety is paramount in therapeutic protein production. Regulatory agencies like the FDA require products to be free of harmful contaminants. E. coli, while efficient, produces endotoxins that necessitate extra purification. Mammalian cells, although costlier, are preferred for many therapeutics due to their reduced risk of contamination.
- Can you manage the process? Efficient protein production requires tight regulation. Inducible expression systems— such as the T7 promoter in bacteria or AOX1 in yeast— allow on-demand control, minimizing stress on host cells and maximizing yield when needed.
- Will the protein fold properly? Function depends on proper folding. Some hosts, like yeast, naturally produce chaperone proteins that assist in folding. In contrast, bacterial hosts often produce misfolded or insoluble proteins, requiring additional steps to recover functional forms.
These factors guide the selection of a host system that matches your protein’s structural needs and aligns with your production goals. For instance, producing a relatively simple protein like insulin in E. coli is efficient and cost-effective due to the bacterium’s rapid growth and high yield capabilities. In contrast, complex therapeutic proteins such as the monoclonal antibody trastuzumab— used in breast cancer treatment— require precise PTMs like glycosylation, which can only be accurately performed in mammalian cells.
Comparing Prokaryotic and Eukaryotic Expression Systems
Let’s explore the two main types of host systems for recombinant protein production: prokaryotic (e.g., bacteria like E. coli) and eukaryotic (e.g., yeast or mammalian cells). Each system comes with its own strengths and limitations, much like choosing between a fast motorcycle and a fully equipped SUV. The choice depends on your destination and what you need to carry (Table 9.1, Figure 9.2).
Table 9.1: Comparison of Prokaryotic and Eukaryotic Expression Systems. CC-BY-SA-4.0, adapted from OpenStax Biology 2e.
| Feature | Prokaryotic (E. coli) | Eukaryotic (Yeast, Mammalian) |
|---|---|---|
| Growth Speed | Super-fast (20–30 min doubling) | Slower (2 hours to days) |
| Cost | Cheap ($0.1–1/L for media) | Pricey ($1–50/L for media) |
| Protein Modifications | Basic (no glycosylation) | Fancy (glycosylation, disulfide bonds) |
| Protein Yield | High (1–5 g/L) | Moderate to Low (0.1–10 g/L) |
| Genetic Engineering | Easy-peasy (simple plasmids) | Trickier (needs advanced tools) |
| Best For | Industrial enzymes, simple drugs | Complex drugs, vaccines |
| Scalability | Great for big bioreactors | Okay, but a more complex setup |
Prokaryotic systems:
Bacterium like E. coli is favored for many protein production applications due to their rapid growth, low cost, and ease of genetic manipulation. With a doubling time of 20–30 minutes, E. coli cultures can reach high densities within hours, making them ideal for large-scale bioreactor use. Media costs are minimal; Luria-Bertani (LB) broth, for instance, costs $0.1–$1 per liter, and simple fermenters further reduce operational expenses. The molecular toolkit for E. coli is extensive, featuring plasmids like the pET vectors series and powerful promoters such as T7, enabling protein expression levels of 1–5 grams per liter. Proteins such as insulin or human growth hormone, which do not require complex PTMs, are well-suited for production in this system. However, E. coli has important limitations. It lacks the cellular machinery for PTMs such as glycosylation, which makes it unsuitable for complex proteins such as monoclonal antibodies, which rely on specific sugar patterns for their function and stability. In addition, proteins expressed in E. coli often form inclusion bodies, insoluble aggregates of misfolded protein that require labor-intensive refolding steps, which can reduce yields by 20–50%. Another problem is the production of endotoxins, which must be rigorously removed in the production of therapeutic proteins for human use. Despite these challenges, E. coli remains an excellent choice for the production of industrial enzymes (e.g., proteases for detergents) and simple therapeutic proteins where speed, scalability, and cost-effectiveness are top priorities.
Eukaryotic systems:
When bacterial systems such as E. coli cannot fulfill the requirements of complex protein expression, eukaryotic systems are used that have the ability to perform key PTMs. These hosts include yeast (Saccharomyces cerevisia, Pichia pastoris), filamentous fungi (Aspergillus niger), and mammalian cells such as Chinese Hamster Ovary (CHO) and HEK293 cells. They are particularly valuable for the production of proteins that require proper folding, disulfide bond formation, and glycosylation - properties that are critical for therapeutic functionality.
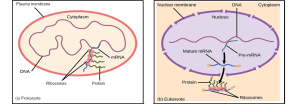
Yeasts, for example, offer a good balance between simplicity and sophistication. P. pastoris is an excellent plant that can produce proteins in high yield with its methanol-inducible AOX1 promoter. It can churn out up to 5–10 grams per liter of recombinant protein, including hepatitis B surface antigens, under high-density fermentation. While yeast glycosylation patterns differ from those of humans, often high-mannose types, they are still beneficial for many applications. The cost is moderate (media ~$1–5/L) and doubling times of 1.5–2 hours make it significantly slower than E. coli, but manageable. Filamentous fungi like A. niger are also used industrially to produce large quantities of enzymes (e.g., amylases, cellulases), with the added benefit of secretion, which simplifies downstream purification.
Mammalian cells, on the other hand, are the gold standard when human-like PTMs are critical, especially for biopharmaceuticals such as monoclonal antibodies (e.g., rituximab for lymphoma or trastuzumab for breast cancer). CHO cells are widely used in industry to produce recombinant proteins. They produce proteins with appropriate glycosylation, folding, and bioactivity. However, this comes at a price: yields typically range from 1–5 g/L, culture media costs can soar to $5–50 per liter, and doubling times extend to 20–30 hours. Despite the costs, mammalian systems minimize misfolding and inclusion body formation due to their internal chaperone systems and advanced protein processing capabilities. Genetically engineering eukaryotic hosts is also more complex. Unlike bacteria, their larger genomes and intricate regulation demand advanced tools like CRISPR/Cas9 or recombinase-mediated cassette exchange. But the payoff is clear when you're producing high-value therapeutics where function, structure, and safety are paramount for medical applications.
9.2 Expression Systems for Recombinant Proteins
Bacterial Systems
Bacterial systems are the reliable workhorses of recombinant protein production, akin to a dependable pickup truck that delivers speed and cost-efficiency. Among these, E. coli stands out as the most widely used host due to its rapid growth, low cultivation costs, and genetic tractability. Under optimal conditions, E. coli can double every 20 to 30 minutes, allowing a culture to grow from a few cells to a high-density biomass capable of producing significant amounts of recombinant protein in less than 24 hours. This makes it ideal for applications requiring large protein quantities on short timelines.
One of the key advantages of bacterial systems is their affordability. Culture media such as Luria-Bertani (LB) or terrific broth are inexpensive, typically costing between $0.10 and $1 per liter, and bacterial fermentation can be performed using relatively simple and low-maintenance bioreactor setups. Additionally, E. coli is genetically well-characterized, supported by a comprehensive suite of molecular tools, including plasmid systems like pET and tightly regulated promoters such as T7 or lac. With engineered strains like BL21(DE3), recombinant protein yields can reach 1–5 grams per liter, making E. coli particularly attractive for producing industrial enzymes (e.g., amylases, proteases) and therapeutic proteins like insulin.
Indeed, the impact of bacterial systems is historic recombinant human insulin, first commercialized in the early 1980s using E. coli, which replaced animal-derived insulin and revolutionized diabetes care through scalable, safer, and more consistent production. However, bacterial hosts are not suitable for all protein types. Their main limitation lies in the absence of eukaryotic cellular machinery for PTMs, such as glycosylation, phosphorylation, and proper disulfide bond formation. These modifications are essential for the structural integrity, biological activity, and pharmacokinetics of many human proteins, especially therapeutic antibodies. For example, monoclonal antibodies produced in E. coli lack the necessary glycan structures to engage immune effector functions, rendering them ineffective for clinical use.
Another challenge is protein misfolding. Overexpression in bacterial systems often overwhelms the cell’s folding capacity, leading to the formation of inclusion bodies, insoluble aggregates of misfolded proteins. Recovering functional protein from these inclusion bodies requires solubilization with denaturants like urea or guanidine hydrochloride, followed by refolding protocols that are time-consuming and often result in 30–50% loss of active protein. Furthermore, bacterial endotoxins, particularly lipopolysaccharides (LPS) in E. coli, pose serious safety concerns for therapeutic applications. These contaminants must be rigorously removed to meet regulatory standards (e.g., FDA, EMA), which increases downstream processing complexity and sometimes offsets the economic benefits of using bacterial systems.
In summary, bacterial hosts are ideal for simple, high-yield protein production, particularly when speed and cost-efficiency are paramount. However, for complex proteins requiring human-like modifications, other expression systems such as yeast, fungi, or mammalian cells are better suited, highlighting the importance of aligning host selection with the biochemical and therapeutic demands of the target protein.
Common Bacterial Hosts
Several bacterial hosts stand out for their unique strengths:
- E. coli: Considered the gold standard for recombinant protein production. It is widely used to produce proteins ranging from insulin to green fluorescent protein (GFP). Strains like BL21(DE3) optimized for T7 promoters offer high expression yields, making them indispensable in both research and industrial applications.
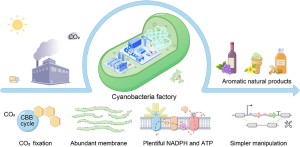
- Cyanobacteria: Photosynthetic bacteria such as Synechococcus and Spirulina are gaining traction as eco-friendly hosts. They harness sunlight to drive protein synthesis, reducing energy inputs for producing enzymes used in biofuels or sustainable chemicals. Figure 9.3 illustrates how cyanobacteria are leveraged for sustainable recombinant protein production.
- Pseudomonas fluorescens: Known for its ability to secrete high levels of soluble proteins, including vaccine antigens and therapeutic peptides, this host can achieve yields up to 2 g/L. It produces fewer inclusion bodies than E. coli, simplifying downstream purification and making it a preferred choice in biopharmaceutical manufacturing.
Applications
Bacterial systems are the backbone of industrial enzyme production, favored for their high yields and cost-effectiveness. Enzymes such as amylases are widely used to break down starches in food processing, while proteases enhance stain removal in laundry detergents. In the pharmaceutical sector, bacteria efficiently produce simple therapeutic proteins like insulin or interferons, with recombinant insulin revolutionizing diabetes care by ensuring a consistent, affordable supply. However, bacterial hosts are generally unsuitable for producing complex proteins such as antibodies or glycoproteins, which require post-PTM, which only eukaryotic systems can perform. In a research setting, E. coli remains a go-to organism for expressing proteins like GFP in fluorescence studies or enzymes for structural biology, owing to its ease of use and high productivity.
Strategies for Improvement
To address the inherent limitations of bacterial expression systems, scientists have developed several innovative strategies to enhance protein solubility and mimic PTMs. Co-expression of molecular chaperones such as GroEL, GroES, and DnaK supports proper protein folding by preventing misfolding and aggregation, often boosting soluble yields by 30–50%. For instance, co-expressing GroEL with an antibody fragment in E. coli reduced inclusion body formation by nearly half, significantly simplifying downstream purification. Another effective approach is lowering the culture temperature to 16–18°C, which slows translation and provides more time for proteins to fold correctly, thereby reducing the formation of insoluble aggregates. Fusion tags such as glutathione S-transferase (GST) or maltose-binding protein (MBP) are frequently used to improve solubility and stabilize proteins during expression and purification. Additionally, signal peptides like PelB can direct proteins to the periplasmic space, a less crowded environment that promotes proper folding, minimizes aggregation, and reduces endotoxin contamination, an important consideration for biopharmaceutical applications. Codon optimization further enhances expression by replacing rare codons in heterologous genes with synonymous codons preferred by E. coli, often improving translation efficiency by up to 40%. For example, codon optimization of a gene encoding a human vaccine antigen led to a two-fold increase in expression levels. To approximate PTMs such as glycosylation, researchers have engineered E. coli strains with heterologous pathways derived from organisms like Campylobacter jejuni, enabling the addition of simple sugar moieties to recombinant proteins. Although these engineered systems are not yet as efficient as eukaryotic hosts in producing fully glycosylated proteins, continuous advancements are rapidly closing the performance gap, offering new possibilities for cost-effective bacterial production of therapeutic proteins.
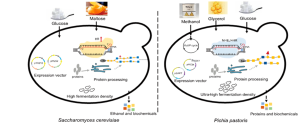
Yeast Systems
Yeast systems offer a versatile platform for recombinant protein production, bridging the gap between the simplicity of bacterial hosts and the advanced PTM capabilities of mammalian cells. Yeasts can carry out important PTMs such as glycosylation and disulfide bond formation, which enable the production of complex biologics such as antibodies, hormones, and vaccine antigens. Among them, S. cerevisiae and P. pastoris (recently reclassified as Komagataella phaffii) are the most commonly used. S. cerevisiae, commonly known as baker’s yeast, is preferred in academic research due to its well-characterized genetics and ease of manipulation. Inducible promoters such as GAL1 allow fine control of gene expression, making it suitable for the production of proteins such as insulin, human serum albumin, or green fluorescent protein (GFP) on a laboratory scale. However, the yield of recombinant proteins is relatively modest (typically 0.5–2 g/L), and the secretion of numerous endogenous proteins can complicate downstream purification, often increasing costs by 20–30%.
In contrast, P. pastoris is optimized for industrial applications. It is characterized by high fermentation density and utilizes the methanol-inducible AOX1 promoter for strong, regulated expression, achieving yields of up to 10 g/L in fed-batch systems. The minimal background secretion of proteins simplifies purification and can reduce downstream processing costs by up to 50 % compared to S. cerevisiae. P. pastoris is often used for the production of therapeutic proteins, such as hepatitis B surface antigen, and enzymes, such as phytases, for animal nutrition. Despite slower growth rates (doubling every ~2 hours) and slightly higher media costs ($1–$5 per liter) than bacteria, yeast systems remain attractive for both research and industrial use because of their ability to produce functionally active, glycosylated proteins. Figure 9.4 compares the strengths of S. cerevisiae and P. pastoris, and illustrates their respective roles in small-scale research and large-scale bioproduction.
PTMs and Glycosylation Patterns
Yeast systems are particularly valued for their ability to perform PTMs that bacterial hosts lack, including N- and O-linked glycosylation and disulfide bond formation. These modifications are critical for the structural integrity, biological activity, and therapeutic efficacy of complex proteins such as antibodies, hormones, and cytokines. Glycosylation, for example, increases the half-life of therapeutic proteins such as erythropoietin in the bloodstream and ensures that they remain stable and functional. The formation of disulfide bonds is also essential for correct protein folding, as is the case with insulin and many immunoglobulins. However, a major limitation of yeast-based systems lies in their glycosylation patterns. Species such as S. cerevisiae and P. pastoris typically add glycans with high mannose content, which differ significantly from the complex, sialylated glycan structures in human cells. These non-human glycosylation patterns can impair the efficacy of biopharmaceuticals and, in some cases, trigger unwanted immune responses.
To overcome these challenges, researchers have engineered glycoengineering yeast strains - in particular, P. pastoris that express human glycosylation enzymes such as mannosidases and glycosyltransferases. These modified strains can produce glycoproteins with more human-like glycan profiles, improving their pharmacokinetic properties and reducing immunogenicity. Although the full humanization of glycosylation in yeast is not yet complete, significant progress has already been made. Several antibody fragments and therapeutic enzymes produced using glycosylated yeasts show improved therapeutic potential. These advances make yeast an effective mediator between the speed and simplicity of bacterial systems and the complex post-translational capabilities of mammalian cells.
Large-Scale Fermentation Strategies
P. pastoris is excellently suited for large-scale fermentation and achieves cell densities of over 100 grams per liter (dry weight) in bioreactors. Its methanol-inducible AOX1 promoter enables precise control of gene expression. However, the methanol content must be carefully regulated to avoid toxicity, which can inhibit growth. Optimal bioreactor conditions include high oxygen transfer rates (100–200 mmol/L/h) and a stable pH range of 6.0 to 7.0, which are critical for maintaining cell health and productivity.
Fed-batch fermentation, where nutrients and methanol are gradually fed, can boost yields by 20–30% compared to batch cultures. Under optimized fed-batch conditions with oxygen sparging and pH control, P. pastoris can produce insulin at concentrations of 5 to 10 grams per liter, making it a leading platform for therapeutic protein production. In contrast, S. cerevisiae is less suited for high-density fermentation due to lower yields but remains a reliable choice for simpler fermentations in research settings, such as expressing GFP for structural biology. Advances in bioreactor technology, such as automated nutrient feeding and real-time monitoring of dissolved oxygen and pH, have further enhanced yeast performance, solidifying their role as essential workhorses in industrial biotechnology for enzyme and drug manufacturing.
Filamentous Fungi
Filamentous fungi such as A. niger and Trichoderma reesei serve as industrial workhorses in recombinant protein production, known for secreting large quantities of extracellular enzymes with yields reaching 20–30 grams per liter. These organisms are particularly valuable for applications demanding high protein output at low cost. Their filamentous hyphal structure supports the dense accumulation of biomass, but also increases the viscosity of the broth, so that bioreactors with improved stirring and oxygen transport capabilities are required. Growth rates are slower than bacteria but faster than mammalian cells, providing a practical trade-off for cost-effective scale-up. Media costs are relatively moderate, typically ranging from $1 to $5 per liter. A major advantage of filamentous fungi is their ability to secrete proteins directly into the culture medium, which simplifies downstream processing and can reduce purification costs by up to 30% compared to intracellular expression systems. However, the non-human glycosylation patterns limit their suitability for the production of therapeutic proteins, as these modifications can trigger immune reactions. In addition, high endogenous protease activity can degrade recombinant proteins if not properly controlled. Despite these challenges, filamentous fungi remain important platforms for the production of industrial enzymes, organic acids, and bio-based materials on a commercial scale.
Use of Aspergillus and Trichoderma
A. niger is widely known for the production of enzymes such as glucoamylase, which plays a key role in the processing of starch for both food and biofuel production. Under optimized conditions, the yield of glucoamylase can be up to 25 grams per liter. This enzyme converts starch into simple sugars, enabling the production of high fructose corn syrup and ethanol. T. reesei, on the other hand, is the most important industrial source of cellulases, which break down plant biomass into fermentable sugars, a critical step in the conversion of lignocellulose into bioethanol. Both fungi rely on strong promoters, such as the cbh1 promoter in T. reesei, to drive high-level expression of recombinant proteins. Although filamentous fungi are less commonly used for the production of therapeutic proteins due to their non-human PTMs, they are being explored for applications where yield and efficiency of secretion outweigh the need for human-like glycosylation. For example, A. niger has been used to produce vaccine antigens from fungi, taking advantage of its robust secretion pathways to streamline production and reduce purification costs.
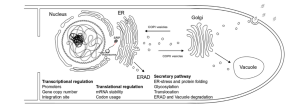
Secretion of High-Yield Extracellular Proteins
Fungi are naturally efficient secretion systems, equipped with special pathways that transport proteins from the cell interior directly into the culture medium. This natural advantage minimizes intracellular accumulation, simplifies further processing, and significantly reduces purification costs. , A. niger, for example, secretes amylases directly into the fermentation broth, enabling simple protein recovery by filtration, unlike bacterial systems that often require cell lysis. Figure 9.5 shows the pathway of protein synthesis and secretion in A. niger, and highlights potential bottlenecks such as transcription, translation, endoplasmic reticulum (ER) stress, glycosylation, and vesicle transport, all of which can affect the efficiency of recombinant protein production. The robust cell walls of filamentous fungi also allow them to withstand the mechanical stresses of high-density fermentation, making them well-suited for large bioreactors with volumes of 1,000 to 10,000 liters. This scalability is critical for industrial enzyme production, especially in sectors such as food processing and bioenergy, where high production volumes are essential.
Genetic Modification Approaches
Genetic engineering plays a key role in improving the performance of fungi. Homologous recombination can be used to enhance promoters or delete protease genes such as pepA, reducing protein degradation by 30–40%. The knockout of pepA in A. niger, for example, has increased glucoamylase yield by preventing degradation of the enzyme during fermentation. CRISPR/Cas9 enables precise interventions, such as upregulation of the cbh1 promoter in T. reesei, which doubled cellulase production for biofuel production. RNA interference (RNAi) increases yields by up to 20 % by silencing genes that compete for cellular resources. These tools make fungi more efficient platforms for both industrial enzymes and new therapeutic applications.
Cell-Free Systems
Cell-free systems function like ready-to-use protein synthesis kits that use cell extracts rich in ribosomes, tRNAs, and enzymes instead of living cells. They enable rapid protein production within hours and offer flexibility in the expression of proteins that are difficult to produce in cells. Reaction conditions can be easily adjusted without concerns about cell viability, making them ideal for quick testing or specialized applications. However, they are expensive ($10–$50/mL), harder to scale, and yield less protein (0.1–2 mg/mL) than systems like E. coli or yeast, though still sufficient for research, prototyping, and structural studies.
Advantages of Cell-Free Protein Synthesis
The speed of cell-free protein synthesis is a major advantage, enabling protein production in hours rather than days. This makes it especially useful for producing proteins that are toxic to living cells, such as antimicrobial peptides, or difficult to express, like membrane proteins. In fact, cell-free systems have become a preferred platform for synthesizing membrane proteins by incorporating lipid nanodiscs, detergents, or liposomes directly into the reaction. Their open format allows precise addition of cofactors, isotope-labeled amino acids, or non-natural amino acids, making them ideal for applications like NMR spectroscopy, protein engineering, and structural biology. For instance, incorporating ^15N- or ^13C-labeled amino acids directly into the mix simplifies production of labeled proteins for high-resolution structural studies. Cell-free systems also support high-throughput workflows, enabling simultaneous screening of hundreds of protein variants, an asset for enzyme engineering and drug discovery.
Types of Cell-Free Systems
- Bacterial Extracts (e.g., E. coli): These are the most cost-effective and high-yield cell-free systems, capable of producing 1–2 milligrams of protein per milliliter for simple proteins like GFP. However, they lack PTM capabilities, limiting their use for complex eukaryotic proteins. They are ideal for producing research reagents and industrial enzymes.
- Wheat Germ Extracts: Derived from plant embryos, these systems support proper eukaryotic
protein folding and enable limited PTMs such as phosphorylation. They are commonly used for plant proteins and basic research applications where moderate complexity is required.
- Insect Extracts (e.g., Sf9): These extracts provide some eukaryotic PTMs, including basic glycosylation, and are suitable for producing complex research proteins, vaccine antigens, and other applications that benefit from limited PTM capability.
- Mammalian Extracts (e.g., CHO or HEK293): Offering the most advanced PTMs such as complex glycosylation and disulfide bond formation these extracts are ideal for producing therapeutic proteins, including antibody fragments. However, they are the most expensive option and are generally reserved for specialized or clinical-grade applications.
Applications
Cell-free systems are highly suited for rapid prototyping tasks such as screening vaccine candidates or evaluating enzyme variants for industrial applications. They play a critical role in structural biology, enabling the production of isotope-labeled proteins for NMR spectroscopy or X-ray crystallography. For instance, E. coli extracts are commonly used to produce labeled GFP for fluorescence studies, while mammalian extracts can generate glycosylated antibody fragments for early-stage therapeutic screening. These systems are also ideal for high-throughput workflows, allowing researchers to test hundreds of protein mutants in parallel accelerating drug discovery and enzyme engineering for applications like biofuel production. Importantly, cell-free platforms excel in synthesizing membrane proteins, which are often difficult to express in living cells. This makes them especially valuable for studying complex drug targets such as G-protein-coupled receptors (GPCRs) and ion channels.
9.3 Strategies for Enhancing Protein Expression
CRISPR-based methods for bacteriophage-resistant strains for large-scale production.
Molecular chaperones act as essential helpers in protein folding, guiding newly synthesized proteins to achieve their correct three-dimensional structures and preventing aggregation into inactive inclusion bodies. In bacterial systems like E. coli, rapid or foreign protein expression often overwhelms the folding machinery, leading to misfolded proteins. Key chaperones such as DnaK, which bind exposed to hydrophobic regions, and the GroEL/GroES complex, which provides a protected folding chamber, assist in proper folding using ATP-driven mechanisms. Co-expressing these chaperones alongside target proteins can significantly increase soluble yields; for example, GroEL/GroES co-expression with recombinant antibody fragments can reduce inclusion bodies by half. In yeast systems like P. pastoris, chaperones such as BiP facilitate the folding and secretion of complex proteins like insulin, enhancing extracellular yields. Typically, chaperone genes are introduced on separate plasmids or integrated into host genomes under inducible promoters (e.g., lac or AOX1), with expression levels optimized to avoid cellular stress. This strategy improves production of industrial enzymes and therapeutics, where solubility directly affects function, cost, and downstream processing. Additionally, chaperone co-expression supports high-throughput screening by enabling rapid access to soluble proteins for testing.
Applications and Examples
Chaperone co-expression has revolutionized protein production in practice. For example, producing human growth hormone in E. coli typically results in inclusion bodies, but co-expressing GroEL/GroES significantly increases soluble protein yields, reducing purification costs. In yeast, overexpressing BiP improves the secretion of monoclonal antibody fragments, vital for cancer therapies. These successes demonstrate why chaperones are a standard tool in biotech labs, enabling the efficient production of challenging proteins.
Mutation Strategies to Improve Protein Expression
Mutation strategies are like tweaking a recipe to perfect a dish, adjusting the protein or host to improve expression and function (Figure 9.6). When a protein expresses poorly or misfolds, scientists use mutagenesis to introduce targeted or random gene changes to find better-performing variants. These approaches are especially useful in hosts like E. coli or yeast, where high expression often leads to misfolding or toxicity.
Site-directed mutagenesis involves changing specific amino acids to enhance stability or solubility. Replacing a hydrophobic residue with a polar residue on the protein surface, for example, can reduce aggregation and increase the soluble yield by 20–30%. This method is based on detailed structural knowledge, often from X-ray crystallography, and is therefore precise but time-consuming. It is often used for enzymes where subtle changes have a major impact.
Random mutagenesis introduces widespread, random genetic changes using methods such as error-prone PCR. The resulting variants are analyzed for improved properties such as higher expression or activity. For example, random mutagenesis of a lipase gene in E. coli resulted in a variant with 50% higher activity, which is ideal for industrial detergents. High-throughput screening enables the rapid testing of thousands of variants, making this approach very powerful for the discovery of improved proteins
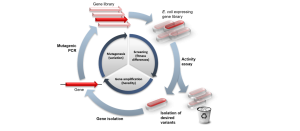
Directed evolution builds on random mutagenesis by mimicking natural selection. Scientists create a diverse library of mutated genes, express them in a host, and select the best performers based on yield or function. This cycle of mutation, screening, and selection is repeated multiple times to progressively improve the protein. Directed evolution has revolutionized enzyme production, for example, evolving cellulase in Trichoderma doubled its efficiency for biofuel applications. ALE focuses on improving the host organism rather than the protein itself. By culturing cells under selective pressures, such as high inducer concentrations or nutrient limitation, the strains develop improved protein production capabilities. For example, ALE has optimized the cellular machinery in E. coli and increased GFP yield two- to threefold. The combination of ALE with genomic analysis helps to identify beneficial mutations and is therefore a powerful strategy for the development of robust industrial strains.
Engineered Expression Systems
Engineered expression systems function like precision tools in the biotechnology toolbox, designed to improve protein production by optimizing the host’s genetic and expression machinery. In bacterial systems, particularly in E. coli, these platforms help overcome challenges such as low yield, protein misfolding, degradation, and contamination during large-scale production. By adapting host strains and expression vectors, researchers have developed robust systems for efficient protein synthesis in research, therapeutic development, and industrial applications. This section introduces several specialized systems, including the pET system, Rosetta strains, protease-deficient strains, Origami strains, and bacteriophage-resistant strains, each developed to overcome specific hurdles in recombinant protein expression.
pET System for Protein Overexpression
The pET system is a widely used and powerful platform for protein production in E. coli, and is often compared to a high-performance engine designed for both speed and precision. At the heart of the system is the T7 promoter, a powerful and highly specific regulatory element derived from the T7 bacteriophage that enables high levels of expression, often accounting for up to 50% of the total protein content of the host cell.
Figure 9.7 shows the main components of the pET system in E. coli, including the T7 promoter, the lac operator, and the IPTG-inducible expression mechanism. In strains such as BL21(DE3), the T7 RNA polymerase gene is integrated into the chromosome under the control of a lac promoter. This configuration allows for precise control of protein expression: the system remains inactive until it is inhibited by isopropyl
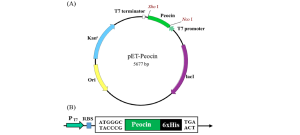
β-D-1-thiogalactopyranoside (IPTG), minimizing cellular stress and toxicity during cell growth. The pET system's versatility makes it ideal for expressing a broad range of proteins, from industrial enzymes like amylases to therapeutic proteins such as insulin. In the production of human growth hormone with this system, yields of up to 3 grams per liter can be achieved under optimized conditions, demonstrating its effectiveness for large-scale production of therapeutic proteins.
Features and Advantages
The pET system is characterized by several key features that make it the gold standard for high-level recombinant protein expression in E. coli. Its cornerstone is the powerful T7 promoter, which drives transcription much more strongly than most native bacterial promoters, enabling efficient expression of target proteins. The modular plasmid design of the system allows easy insertion of genes of interest, enabling researchers to quickly customize the vector to different experimental requirements. A wide range of pET vectors is available with various affinity- and solubility-enhancing tags, such as His tags for rapid purification by metal affinity chromatography, or GST and MBP tags to improve solubility and folding, which greatly simplifies downstream processing and improves overall protein yield and functionality.
One of the greatest strengths of the pET system is its tightly controlled expression. The use of the lac operator, in combination with the inducible T7 RNA polymerase system, ensures minimal background or “leaky” expression in the absence of inducers such as IPTG. This precise control is particularly beneficial when toxic or unstable proteins are expressed, as it helps to maintain cell viability and reduce the risk of premature degradation or misfolding. In addition, the system is highly adaptable to a range of E. coli host strains. , BL21(DE3), for example, is commonly used for general expression and lacks key proteases, while Rosetta strains provide additional tRNAs for efficient translation of eukaryotic genes. Other strains, such as Origami, are engineered to support the formation of disulfide bonds in the cytoplasm, expanding the range of proteins that can be successfully expressed.
Optimization of Expression Conditions
To maximize protein yield in the pET system, the expression conditions are carefully optimized to achieve a balance between productivity and cell health. The IPTG concentration is usually between 0.1 and 1 mM, although lower concentrations are often preferred to avoid overwhelming the host’s protein folding machinery, which can lead to the formation of insoluble inclusion bodies. In addition, the temperature in the culture is lowered to 16–25 °C to slow down protein synthesis and allow more time for proper folding. This strategy has been shown to increase the solubility of proteins such as GFP by 30–40%.
Autoinduction media offer an alternative approach for large-scale expression. They allow the culture to self-induce protein production once glucose is depleted. In addition, co-expression of molecular chaperones such as GroEL and GroES can promote proper protein folding and significantly reduce aggregation.
For example, optimizing IPTG concentration to 0.5 mM and lowering the growth temperature to 18°C led to a twofold increase in the yield of a recombinant vaccine antigen expressed using the pET system. Such fine-tuning strategies are crucial for improving the efficiency and scalability of protein production for both research and industrial applications.
Rosetta (E. coli) Strain Engineering for Rare tRNA
Rosetta E. coli strains function as molecular translators, enhancing the host’s ability to express genes from foreign sources, particularly AT-rich or codon-biased eukaryotic genes. Many genes from organisms such as humans or plants contain codons that are rare in E. coli, for example, AGA or AGG for arginine, which can stall ribosomes during translation and significantly reduce protein yields. Rosetta strains address this limitation by supplying additional tRNAs for several rare codons, thereby improving translational efficiency and boosting protein expression by two- to threefold. Figure 9.8 highlights a variety of E. coli strains engineered for high-level protein expression under the control of the T7 promoter, including Rosetta strains optimized for rare codon usage. For instance, expressing a human antibody fragment in Rosetta E. coli increased yield from 0.5 to 1.5 grams per liter, a threefold improvement representing a major advancement in recombinant therapeutic protein production.
Enhancing Expression of AT-Rich Genes
AT-rich genes, particularly those from eukaryotic sources, often pose challenges in E. coli due to the frequent use of codons that are rare in the bacterial genome. When these rare codons are encountered, the limited availability of corresponding tRNAs can lead to ribosome stalling, incomplete translation, and the production of truncated or misfolded proteins. This bottleneck is especially problematic when expressing structurally complex or functionally critical proteins, such as membrane receptors or multi-domain enzymes, where even modest expression levels are essential for drug screening, functional assays, or structural biology. Rosetta strains address this issue by harboring plasmids like pRARE, which supply tRNAs for seven rare codons (including AGA, AGG, AUA, CUA, CCC, GGA, and CGG). By supplementing the host’s tRNA pool, these strains facilitate smooth and continuous translation of heterologous genes, significantly improving both protein yield and quality. This capability makes Rosetta strains indispensable tools for expressing challenging eukaryotic proteins in bacterial systems.
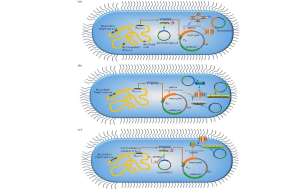
Codon Optimization and tRNA Supplementation
Codon optimization further enhances the performance of Rosetta strains by modifying the gene sequence to use codons preferred by E. coli, thereby improving translational efficiency and protein yield often by up to 40%. For instance, codon optimization of a human gene encoding a vaccine antigen resulted in a twofold increase in expression in E. coli. Advanced software tools, such as GeneOptimizer, facilitate this process by analyzing host-specific codon usage patterns, GC content, mRNA stability, and other parameters to design optimized gene sequences tailored to the expression system.
When combined with Rosetta strains, which supply rare tRNAs to overcome codon bias, codon optimization creates a synergistic effect that maximizes protein production. This dual strategy is particularly effective for the expression of AT-rich genes from organisms such as humans or Plasmodium species (malaria parasites), where rare codon usage would otherwise limit expression. This enables the production of complex target genes such as malaria antigens with high yields, making the Rosetta strains indispensable tools in biotechnology and vaccine development.
Protease-Deficient Strains
Proteases in E. coli function like overactive cleaning columns, often degrading newly synthesized recombinant proteins before they can be properly folded, enriched, or harvested. Endogenous proteases such as Lon and OmpT are particularly problematic, as they target misfolded or unstable proteins, commonly found during high expression. This degradation not only reduces the yield but also makes purification and further processing more difficult. For this reason, protease-deficient strains have been developed that lack key proteolytic enzymes, thereby stabilizing the recombinant proteins and improving overall expression efficiency. These strains are especially valuable for the production of sensitive or easily degradable proteins, such as antibody fragments, cytokines, and growth factors, where even partial degradation can impair biological activity or therapeutic potential. By minimizing proteolysis, these specialized E. coli hosts have become important tools to increase the yield and maintain the structural integrity of challenging recombinant proteins.
Impact of Protease Activity
Proteases in E. coli often target exposed or misfolded regions of recombinant proteins, especially those overexpressed under strong promoters. This proteolytic activity can significantly reduce yield and compromise protein quality. For example, expression of GFP in wild-type E. coli can result in 20–30% of the protein being degraded, leading to lower yield and higher purification costs. The effects are even more critical in the production of therapeutic proteins, where even a small amount of degradation can render the product biologically inactive or trigger unwanted immune reactions. In such cases, control of protease activity is critical to ensure the integrity, safety, and efficacy of the final product.
Engineering Strains for Protease Knockout Mutations
Protease-deficient strains, such as BL21(DE3) derivatives lacking the lon and ompT genes, can reduce protein degradation by 30 to 40%. These gene knockouts are usually generated using precise genome editing methods such as homologous recombination or CRISPR/Cas9, which enable targeted deletion of specific protease genes. The use of a protease-deficient E. coli strain, for example, led to a 35% increase in the yield of a recombinant enzyme, making it highly suitable for industrial production. Due to their ability to conserve unstable proteins, these strains have become the standard hosts for the expression of sensitive recombinant proteins, ensuring higher yields and higher quality products for applications in drug development, enzyme engineering, and beyond.
Origami Strain for Disulfide Bond Formation
Origami E. coli strains serve as specialized hosts that facilitate the formation of disulfide bonds, which are critical for the stability and function of proteins such as antibodies and insulin. In contrast to wild-type E. coli, whose cytoplasm has a reducing environment that inhibits the formation of disulfide bonds, Origami strains overcome this limitation by genetically modifying the cell’s redox pathways to create a more oxidizing cytoplasm that favors the formation of disulfide bonds.
Enhancing Correct Protein Folding
Origami strains are engineered to lack thioredoxin reductase (trxB) and glutathione reductase (gor), creating an oxidizing cytoplasmic environment that promotes the formation of disulfide bonds. This modification enables complex proteins such as antibody fragments to fold properly, resulting in up to 40% higher yields of functional proteins. For example, the production of a single-chain antibody in Origami E. coli achieved a yield of 1 gram per liter of active protein, whereas expression in standard strains often results in negligible amounts.
Engineering Redox Pathways
By knocking out trxB and pgi, Origami strains shift the cytoplasmic redox balance to mimic the oxidizing environment of the eukaryotic endoplasmic reticulum, which is important for disulfide bond formation. Additional enhancements, such as the overexpression of disulfide isomerases, improve the accuracy and efficiency of disulfide bond formation. These modifications make Origami strains particularly well suited for the expression of proteins with multiple disulfide bonds, such as therapeutic peptides and complex enzymes, which are a key limitation of conventional bacterial expression systems.
Bacteriophage-Resistant Strains
Contamination with bacteriophages poses a serious threat to large-scale bacterial fermentations, comparable to an uninvited guest disrupting a carefully planned event and causing widespread disruption. Phages infect E. coli cells and hijack their machinery to rapidly multiply and eventually lyse the host bacteria, which can lead to a complete breakdown of the culture and disruption of protein production. This not only leads to significant yield losses but also increases downtime and operating costs in industrial bioprocessing. To combat this, bacteriophage-resistant E. coli strains have been developed, often through precise genome editing techniques such as CRISPR/Cas9. These genetically engineered strains carry genetic modifications that prevent phage attachment, entry, or multiplication, providing robust defenses against common phage infections. The use of bacteriophage-resistant strains in industrial bioreactors helps to maintain the stability of cultures, ensure consistent protein yields, and safeguard the overall efficiency of bioproduction processes.
Strategies for Preventing Bacteriophage Contamination
Phage contamination is a major challenge in large-scale fermentation, as even a single viral infection can reduce product yield by 80 to 90 %. Although strict sterilization and hygiene protocols mitigate this risk, they cannot eliminate the danger. To ensure consistent and robust production, genetically modified E. coli strains with built-in phage resistance mechanisms are invaluable. These strains effectively protect cultures in bioreactors with a capacity of 1,000 to 10,000 liters, keeping productivity high and minimizing costly downtime.
Development of CRISPR-Based Resistance Systems
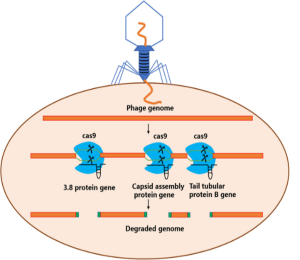
CRISPR/Cas9 systems protect E. coli from phage infections by encoding guide RNAs that instruct the Cas9 nuclease to specifically recognize and cleave viral DNA sequences, and thus prevent phage replication. For example, E. coli strains engineered with CRISPR-based defenses have demonstrated a 90% reduction in phage-related losses during large-scale insulin production. Figure 9.9 illustrates the CRISPR/Cas9 system implemented in BL21 E. coli, programmed to target and cleave the T7 phage genome, enhancing bacteriophage resistance.
These systems can be designed to recognize multiple phage types, providing broad-spectrum protection. When combined with complementary strategies such as modifying bacterial surface receptors to block phage attachment and entry CRISPR-based defenses significantly bolster strain robustness. Together, these innovations make engineered E. coli strains indispensable for maintaining stable, high-yield production in industrial biotechnology.
9.4 Metabolic Engineering for Enhanced Protein Production
Metabolic Pathway Engineering
Metabolic pathway engineering is like fine-tuning a car engine to maximize fuel efficiency and redirecting a cell’s resources to optimize the production of recombinant proteins. By altering the metabolic pathways of host organisms, such as E. coli or P. pastoris, scientists ensure that cellular energy, nutrients, and machinery are prioritized for protein synthesis rather than other competing processes. This approach is crucial in biotechnology because high yields of functional proteins such as insulin or industrial enzymes lead to significant cost savings and improved product quality. Metabolic engineering requires a deep understanding of the cell’s metabolic network, a complex system of interconnected biochemical reactions. Through targeted interventions, such as boosting amino acid biosynthesis, increasing energy production, or down-regulating metabolic pathways that divert resources, researchers can redirect cellular metabolic fluxes to the desired product. The result is a host cell that is transformed into a highly efficient, streamlined factory that can produce proteins in larger quantities and with higher yields.
Rational Design of Metabolic Fluxes
Rational design uses computer models and biochemical insights to optimize metabolic fluxes - the flow of metabolites through cellular metabolic pathways to improve protein production. In E. coli, for example, overexpression of key glycolytic genes such as pgi (phosphoglucose isomerase) increases the generation of ATP and NADPH, vital energy sources that support recombinant protein synthesis. This strategy can increase the yield of enzymes such as amylases by 20 to 30 %. Computational tools such as flux balance analysis (FBA) help to identify the right metabolic pathways and uncover bottlenecks where metabolic intermediates are diverted to competing processes. In P. pastoris, metabolic engineers improve methanol utilization by fine-tuning the expression of AOX1-related genes, directing more carbon flux into protein production rather than biomass accumulation. For example, optimization of methanol metabolism in P. pastoris has increased insulin yield in bioreactor cultures from 5 to 8 grams per liter. These targeted metabolic adjustments require a comprehensive knowledge of host cellular metabolism, often supported by multi-omics data, including genomics and proteomics, to map and prioritize critical metabolic pathways. Rational design also means that gene expression must be carefully balanced, as excessive overexpression can lead to cellular stress, which ultimately reduces growth rates and protein yields.
Eliminating Competing Pathways
Competing metabolic pathways act like leaky pipes, diverting valuable cellular resources away from recombinant protein synthesis. By eliminating or downregulating these metabolic pathways, scientists can effectively “seal" the leaks and release precursors and energy for protein production. In E. coli, for example, silencing genes such as ackA (acetate kinase) reduces the formation of acetate, a wasteful by-product, and redirects carbon flux to amino acid biosynthesis. Figure 9.10 shows how the carbon fluxes of glucose and xylose are redirected in engineered E. coli strains, where the disruption of competing metabolic pathways increases the yield of recombinant proteins. This strategy has been shown to increase GFP production by 25 % in high-performance cultures. Similarly, in yeast, knocking out genes involved in ethanol production, such as ADH1 (alcohol dehydrogenase), directs more glucose into protein synthesis, increasing the yield of therapeutic proteins such as hepatitis B antigens by about 30 %. These gene knockouts are carefully selected to avoid interfering with essential cellular functions, and metabolic models are used to predict their further effects. For example, deletion of ldhA in E. coli to reduce lactate formation improved enzyme production without affecting cell growth, providing an optimal balance that is critical for industrial applications.
Integration of Synthetic Biology Tools
The tools of synthetic biology, including modular genetic circuits and synthetic promoters, provide a versatile toolbox for the development and fine-tuning of individual metabolic pathways. Synthetic promoters provide precise control over gene expression and ensure that key enzymes are produced at optimal levels to maximize metabolic efficiency. For example, synthetic promoters engineered in E. coli have been used to regulate amino acid biosynthesis genes, boosting precursor availability to produce proteins like insulin. Dynamic genetic circuits, such as toggle switches, enable cells to respond adaptively to metabolic demands, preventing overload and maintaining cellular health. In P. pastoris, synthetic biology has enabled the introduction of human-like glycosylation pathways through the incorporation of mammalian enzymes into cassettes, significantly enhancing the quality and therapeutic efficacy of recombinant proteins. Standardized DNA parts, such as those provided by the BioBricks framework, streamline the assembly and optimization of complex synthetic pathways. For instance, engineering a synthetic lysine overproduction pathway in E. coli increased antibody fragment yields by 40% by ensuring an ample supply of key metabolic precursors. Together, these synthetic biology tools make metabolic engineering more precise, scalable, and adaptable across a broad range of protein targets, accelerating advances in biotechnology and therapeutic protein production.
Engineering Methods for Strain Development
Engineering methods for strain development are similar to improving the machinery in a factory to maximize production. These methods transform bacteria, yeasts, or fungi into highly efficient protein factories by precisely modifying their genomes, introducing new genetic elements, or applying adaptive evolution under selective pressure. State-of-the-art tools such as CRISPR/Cas9, recombinant DNA technologies, and directed evolution have revolutionized strain engineering, enabling targeted modifications that improve yield, stability, and scalability. These strategies are essential for the production of a wide range of products - from industrial enzymes to complex therapeutics - and ensure that microbial hosts perform reliably in large bioreactors.
CRISPR/Cas9 Genome Editing for Targeted Modifications
CRISPR/Cas9 works like a molecular scalpel, enabling precise and efficient interventions in a host’s genome to increase protein production. Guided by RNA sequences, the Cas9 enzyme targets specific DNA loci to make cuts that enable the insertion, deletion, or replacement of genes with high accuracy. In E. coli, CRISPR/Cas9 was used to knock out competing metabolic genes such as pta (phosphotransacetylase), redirecting carbon flow and increasing GFP yield by 30%. Figure 9.11 illustrates the knockout of ldhA in E. coli, which similarly redirects metabolic flow to enhance recombinant protein production by up to 30%. carbon flux to enhance recombinant protein production by up to 30%. In P. pastoris, CRISPR-mediated edits of the AOX1 promoter have boosted insulin production by 25% during high-density fermentation. Multiplexed CRISPR systems can simultaneously target multiple genes, streamlining complex strain engineering. For example, editing three metabolic genes in A. niger increased glucoamylase yields by 40%.
Beyond metabolic pathway modifications, CRISPR also overcomes host limitations by enabling the integration of heterologous pathways, for instance, adding glycosylation enzymes to E. coli to facilitate the production of simple glycosylated proteins. Thanks to its precision, versatility, and rapid turnaround, CRISPR/Cas9 has become an indispensable tool in modern strain engineering, reducing development timelines from months to weeks.
Recombinant DNA Technologies for Strain Optimization
Recombinant DNA technologies are fundamental tools in biotechnology, comparable to a master craftsman transforming raw materials into a refined product. In these methods, foreign DNA, such as plasmids or gene cassettes, is introduced into host organisms to improve protein production. In E. coli, plasmids such as pET carry strong promoters (e.g., T7) alongside the target genes, enabling protein yields of 3 to 5 grams per liter for products such as insulin. In yeast, stable expression is achieved by integrating gene cassettes directly into the genome, which is critical for sustainable protein production during long-term fermentation. For example, inserting a cellulase gene under the cbh1 promoter in T. reesei doubled the enzyme output, which is beneficial for biofuel applications. Techniques such as homologous recombination allow the precise insertion of genes at specific genomic locations and offer greater stability than plasmid-based expression systems. In addition, synthetic DNA libraries created by gene synthesis facilitate rapid screening of promoter variants and other regulatory elements to fine-tune expression levels. These recombinant DNA technologies remain essential to construct robust production strains, especially in combination with advanced tools such as CRISPR for genome editing.
Adaptive Evolution Approaches for High-Yield Strains
Adaptive laboratory evolution (ALE) is comparable to training an athlete through repeated challenges, where microbial strains evolve under selective pressure to improve their performance. By culturing cells under conditions that favor high protein production, such as increased induction concentrations or nutrient limitation, scientists select mutants with improved traits. In E. coli, ALE under IPTG-induced stress increased GFP production two- to threefold by optimizing translation efficiency. In P. pastoris, ALE with methanol selection improved AOX1-driven expression and increased antibody fragment yield by 35%. ALE is often combined with genomic analyses to identify beneficial mutations, e.g., those that upregulate amino acid biosynthetic pathways. Continuous culture systems, such as chemostats, maintain selection pressure over several generations and accelerate the evolutionary process. For example, ALE applied to A. niger improved glucoamylase secretion by 30%, demonstrating its value as a strategy for developing industrial strains where maximizing yield is critical.
9.5 Future Perspectives and Emerging Research Areas
The field of recombinant protein production is like a spacecraft charting a course toward uncharted frontiers, propelled by cutting-edge technologies that promise to redefine biotechnology. Breakthroughs in synthetic biology, AI, novel host systems, and sustainable production strategies are revolutionizing how proteins are designed, manufactured, and scaled for medicine, industry, and research. These new innovations aim to overcome persistent challenges, such as high production costs, complex protein modifications, and environmental impacts, while opening up new opportunities such as personalized therapies, sustainable biomaterials, and carbon-neutral bioprocesses. The future of protein production will offer faster, cheaper, and more environmentally friendly solutions through the integration of advanced computational tools, artificial hosts, and green methods. This change has the enormous potential to revolutionize global health, agriculture, and environmental protection on a global scale.
Advances in Synthetic Biology for De novo Protein Design
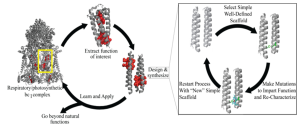
Synthetic biology is like a master architect designing proteins from the ground up, designing molecules with customized structures and functions to meet specific needs. In contrast to traditional approaches, where existing genes are modified, de novo protein design uses advanced computational tools to create entirely new proteins with customized properties such as increased catalytic activity, stability, or binding specificity. Software platforms such as Rosetta and AlphaFold predict how amino acid sequences fold into three-dimensional structures, enabling the development of enzymes that outperform their natural counterparts. Figure 9.12 illustrates the workflow for de novo protein design, from computational modeling to genetic circuit assembly. For example, a synthetic enzyme engineered for biofuel production doubled cellulose breakdown efficiency compared to natural cellulases, a major leap forward for sustainable energy. These tools also allow scientists to model protein interactions with substrates or receptors, optimizing designs for applications like targeted drug delivery, where synthetic antibodies bind cancer cells more effectively than natural ones.
Beyond protein design, synthetic biology harnesses modular genetic circuits and programmable switches that precisely control protein expression. In E. coli, synthetic promoters and riboswitches dynamically regulate gene expression in response to cellular signals such as metabolite levels, maximizing production without compromising cell health. This approach increased insulin yields by 30% by balancing expression with cellular fitness. In P. pastoris, synthetic pathways introduce human-like glycosylation enzymes, including mannosidases and glycosyltransferases, which enable the production of therapeutic proteins such as monoclonal antibodies with improved efficacy in humans. These metabolic pathways are assembled with standardized DNA building blocks such as BioBricks, which accelerate the assembly of complex genetic systems. For example, the development of a synthetic pathway in E. coli to produce novel antimicrobial peptides has doubled yields compared to conventional methods, paving the way for new treatments against antibiotic-resistant infections.
Another exciting area is the development of non-natural proteins with novel functions, such as enzymes that can break down plastics or bind CO₂ to capture carbon. These proteins are developed through a combination of computer-aided design and high-throughput screening, in which thousands of variants are tested to identify the most effective candidates. In yeast, synthetic biology has enabled the production of spider silk proteins that are both stronger than steel and biodegradable, offering sustainable alternatives to petroleum-based materials. These advances extend the potential applications of recombinant proteins beyond medicine and traditional industry to new areas such as environmental remediation and modern materials science. Here, the proteins serve as versatile building blocks for environmentally friendly products such as bioplastics and tissue scaffolds for regenerative medicine. While scaling up these synthetic systems for industrial production remains a challenge, researchers are working to improve their robustness. Pilot studies with optimized synthetic strains have already shown yield increases of 20 to 30 %, which is a promising step towards market maturity.
AI and ML for Predicting Expression Outcomes
AI and ML act as experts that manage the complexity of protein production, predict outcomes, and optimize processes with remarkable precision. These technologies analyze vast data sets from genomics, transcriptomics, proteomics, and fermentation experiments to uncover patterns that improve yield, solubility, and protein function. Machine learning models trained on expression data from hosts such as E. coli or P. pastoris can predict optimal promoter strengths, codon usage, and chaperone quantities, significantly reducing the need for time-consuming laboratory screening. For example, AI-assisted codon optimization for a vaccine antigen in E. coli has increased yields by 40% without the need for repeated trials.
Deep learning algorithms, such as AlphaFold's, predict protein structures at atomic resolution and guide targeted mutations to improve stability or activity. This capability has accelerated enzyme engineering, e.g., through a lipase variant with 50% higher activity optimized for detergent production.
In biopharmaceutical production, AI platforms such as DeepProtein analyze host modifications and predict the impact of strategies such as chaperone co-expression or protease silencing on protein yield. For example, AI-driven adjustment of chaperone concentration in E. coli has increased antibody fragment production by 35%, saving months of experimentation. These tools also enable the optimization of fermentation parameters such as temperature, pH, and nutrient supply in real time. In P. pastoris, AI-driven bioreactor management increased insulin production by 25% through dynamic methanol induction. AI also predicts the toxicity and immunogenicity of proteins, which are crucial for therapeutic safety. Machine learning models have identified immunogenic regions in synthetic antibodies, helping to reduce adverse immune reactions in clinical trials.
The real power of AI lies in the integration of different data streams, from gene sequences to sensor results from bioreactors, providing a holistic view of the production process. New approaches, such as reinforcement learning, are being explored to develop entirely new expression systems and iteratively improve strains based on performance feedback. These innovations have shortened development times from months to weeks and reduced pilot-scale production costs by 15-20%. Despite this progress, challenges remain, such as the need for larger, standardized data sets to improve model training. Joint initiatives to share multi-omics data address this gap and promise even more precision and efficiency in future protein production.
Use of Novel Hosts for Recombinant Protein Production
Novel hosts such as microalgae and plant systems are developing into innovative, environmentally friendly factories for protein production. Microalgae species such as Chlamydomonas reinhardtii and Nannochloropsis use sunlight and CO₂ to drive protein synthesis, significantly reducing energy costs compared to conventional hosts such as E. coli or mammalian cells. These microalgae can produce proteins, including vaccine antigens, in photobioreactors with a yield of 1 to 2 grams per liter at a media cost of only 0.50 US dollars per liter. Their eukaryotic cellular machinery supports complex PTMs such as glycosylation, which are important for therapeutic proteins. For example, C. reinhardtii has been engineered to produce malaria vaccine candidates with glycans compatible with humans - a feat that goes beyond bacterial systems. Advances in genetic engineering, such as CRISPR/Cas9, have further improved the microalgal strains and increased expression and secretion. Recent reports show up to 30% higher yields of antibody fragments.
Plant-based systems such as tobacco (Nicotiana benthamiana), rice, and lettuce offer unparalleled scalability through field or greenhouse cultivation. They produce proteins such as antibodies in quantities of 0.5 to 1 gram per kilogram of plant tissue, using a low-cost agricultural infrastructure. Transient expression with viral vectors derived from the tobacco mosaic virus enables the production of proteins within days, making these systems ideal for rapid-response vaccines. During the Ebola outbreak in 2014, N. benthamiana was used to produce ZMapp antibodies within a few weeks, demonstrating its potential for emergency biopharmaceutical applications. The stable transformation, which integrates genes into the plant genome, enables the long-term production of proteins such as insulin in seeds that can be stored for extended periods of time. Plants also provide human-like glycosylation patterns that improve therapeutic efficacy, although yields generally lag behind those of yeast or fungi, and regulatory approval of plant-derived biologics is a hurdle.
Both microalgae and plant hosts face challenges, such as optimizing expression levels and navigating complex regulatory pathways for therapeutic products. However, their sustainability, which relies on sunlight, CO₂, or existing agricultural systems, makes them very attractive for the production of proteins such as biofuel enzymes or antibodies to improve global health, especially in resource-limited areas. Research is also exploring other novel hosts, including extremophiles and synthetic cells, which promise to further diversify and expand protein production platforms.
Sustainable and Cost-Effective Production Strategies
Sustainability is the green fuel for the future of protein production, with the aim of reducing costs, energy consumption, and environmental impact. A key strategy is to utilize renewable feedstocks such as agricultural waste or lignocellulosic biomass as carbon sources for microbial hosts such as E. coli or A. niger. This approach lowers media costs by 20-30% and reduces dependence on fossil fuel-derived nutrients, significantly reducing the environmental footprint. For example, the use of corn stover as feedstock for E. coli fermentation reduced the cost of amylase production by 25%. Photosynthetic organisms such as microalgae and cyanobacteria produce proteins, including enzymes for biofuels, with near-zero energy input, making them ideal for sustainable bioprocessing.
Continuous fermentation systems, unlike conventional batch methods, ensure consistent production and increase yields by 25% while minimizing waste. For P. pastoris, continuous fermentation with automatic methanol feed improved the efficiency of antibody production and reduced costs by 15%. Recycling bioreactor by-products such as spent media or biomass further reduces waste and costs; for example, recycling yeast media components in S. cerevisiae cultures reduced nutrient costs by 10%. Synthetic biology tools such as biosensors enable real-time monitoring of cell health and dynamically adjust nutrient delivery to optimize resource consumption. In E. coli, biosensors that regulate glucose uptake increase GFP yield by 20% by preventing excessive nutrient consumption.
Process intensification techniques, such as high-density perfusion systems, maximize bioreactor performance and achieve up to 15 grams per liter of therapeutic proteins in P. pastoris. These methods reduce water and energy consumption, and thus meet the goals of green biotechnology. New strategies, such as the co-cultivation of different hosts to share metabolic loads, are also gaining in importance. For example, the co-cultivation of E. coli and S. cerevisiae has increased enzyme yields by 30 % by efficiently sharing the synthesis tasks. Taken together, these sustainable approaches not only reduce production costs but also meet the global demand for environmentally friendly bioprocesses and make protein production more accessible for applications such as affordable vaccines and biodegradable materials.
9.6 Conclusion
Recombinant protein expression has revolutionized biotechnology, transforming small laboratories into global centers for the production of life-saving drugs, industrial enzymes, and research tools. Decades of innovation in host systems, genetic engineering, and process optimization have overcome challenges such as low yields and complex modifications, making protein production more efficient and accessible. New technologies promise even more sustainable, precise, and scalable solutions. E. coli continues to be the backbone of protein production due to its rapid growth and low media costs, producing proteins such as insulin and GFP at 1–5 grams per liter. Strains such as BL21(DE3) paired with pET vectors provide tight expression control, while co-expression of chaperones and codon optimization increase soluble protein yields by 30–50%. Engineered strains that introduce artificial PTMs solve bacterial limitations.
Yeasts such as S. cerevisiae and P. pastoris combine bacterial lightness with eukaryotic PTMs, and achieve up to 10 grams per liter during fermentation. The AOX1 promoter and secretion pathways of P. pastoris cut purification costs in half, and engineered glycosylation improves therapeutic efficacy. Filamentous fungi such as A. niger and T. reesei are excellent at producing enzymes at 20–30 grams per liter for the biofuel and food industries. Genetic tools such as CRISPR/Cas9 and RNA interference increase yields by 20–40%.
Cell-free systems enable rapid protein synthesis for screening and prototyping, with a yield of up to 2 mg/ml, albeit at a higher cost. Metabolic engineering optimizes metabolic pathways to increase yields by 25–30%, while synthetic biology tools fine-tune gene expression in hosts such as P. pastoris. The future lies in synthetic biology developing new proteins for plastic degradation or personalized antibodies, and in AI/ML accelerating strain development, and shortening the timeline from months to weeks. AI-driven optimization of E. coli has increased the yield of vaccine antigens by 40%. New hosts such as microalgae (Chlamydomonas reinhardtii) and plants (Nicotiana benthamiana) provide sustainable, cost-effective platforms to produce proteins with human-tolerated PTMs. Continuous fermentation and renewable raw materials reduce environmental impact and production costs by 15–25%.
Challenges remain, including replicating human-like glycosylation in non-mammalian hosts, scaling novel systems, and regulatory hurdles for plant-based therapies. However, integration of AI, synthetic biology, and green bioprocessing, supported by shared omics data and advanced gene editing, promises to overcome these barriers. This will expand access to affordable biologics, improve industrial bioprocesses, and unlock new applications in sustainable materials and carbon-neutral production transforming global health, industry, and the environment.
Recommended Reading Materials
- OpenStax Biology 2e: https://openstax.org/details/books/biology-2e
- Wikimedia Commons for biotech images: https://commons.wikimedia.org/w/index.php?search=microbial+biotechnology
Resources:
- Rosano, G. L., & Ceccarelli, E. A. (2014). Recombinant protein expression in E. coli: Advances and challenges. Frontiers in Microbiology, 5, 172.
- Demain, A. L., & Vaishnav, P. (2009). Production of recombinant proteins by microbes and higher organisms. Biotechnology Advances, 27(3), 297–306.
- Carlson, E. D., et al. (2012). Cell-free protein synthesis: Applications and challenges. Biotechnology Advances, 30(5), 1185–1194.
- Walsh, G. (2018). Biopharmaceuticals: Biochemistry and Biotechnology (2nd ed.). Wiley.
- Arnold, F. H. (2018). Directed evolution: Bringing new chemistry to life. Angewandte Chemie International Edition, 57(16), 4143–4148.
- Kim, J. Y., et al. (2012). Disulfide bond formation in E. coli : Application to recombinant protein production. Applied Microbiology and Biotechnology, 93(4), 1413–1420.
- Barrangou, R., & Doudna, J. A. (2016). Applications of CRISPR technologies in research and beyond. Nature Biotechnology, 34(9), 933–941.
- OpenStax Biology 2e: https://openstax.org/details/books/biology-2e
- Nielsen, J., & Keasling, J. D. (2016). Engineering cellular metabolism. Cell, 164(6), 1185–1197.
- Lee, S. Y., et al. (2019). Metabolic engineering of microorganisms for biofuels production. Nature Reviews Microbiology, 17(8), 462–475.
- Jumper, J., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589.
- Specht, E. A., & Mayfield, S. P. (2014). Algae-based biopharmaceuticals. Biotechnology Letters, 36(2), 191–197.
- Buyel, J. F., et al. (2017). Plant molecular farming: Opportunities and challenges. Journal of Biotechnology, 260, 1–8.
- Nielsen, J., & Keasling, J. D. (2016). Engineering cellular metabolism. Cell, 164(6), 1185–1197.
