
13 Chapter 13. Microbial uses in environmental biotechnology
Suhaib Ahmad and Abdul Latif Khan
CHAPTER OUTLINE
- Microbial Bioremediation and Environmental Applications
- Microbes in Wastewater Treatment and Heavy Metal Detoxification
- Microbial Biosensors and Energy Production
- Metagenomics and Omics Approaches in Environmental Biotechnology
Learning Objectives
By the end of this chapter, you will be able to:
- Explain how microbes help in environmental applications such as bioremediation, wastewater treatment, and biofuel production.
- Understand the use of microbial tools and biosensors in detecting and managing pollutants.
- Describe metagenomics and other omics approaches for studying environmental microbial communities.
Introduction
Microorganisms play a central role in maintaining environmental balance and driving ecosystem processes. Their metabolic diversity allows them to degrade pollutants, recycle nutrients, detoxify hazardous compounds, and even produce renewable forms of energy. These capabilities make microbes invaluable allies in addressing global challenges such as pollution, climate change, and sustainable resource management. Environmental biotechnology harnesses these microbial functions to develop solutions that are not only effective but also environmentally friendly.
Microbial Bioremediation
Microbial bioremediation refers to the use of microorganisms or their metabolic products to remove pollutants from the environment. In the United States alone, an estimated 200 million tons of hazardous waste are generated each year, originating from industrial spills, illegal dumping, and residues at abandoned manufacturing sites. Traditional cleanup approaches are often costly and disruptive, whereas microbes offer a more efficient and sustainable solution.
Microbes are especially suitable for remediation because of their extraordinary metabolic diversity. They can use a wide range of organic and inorganic compounds as sources of energy or nutrients, breaking down contaminants into harmless byproducts such as carbon dioxide, water, and biomass. These natural processes are cost-effective, particularly when carried out on site, and are generally less damaging to ecosystems compared to chemical or physical treatments (Jaiswal & Shukla, 2020).
Applications of microbial bioremediation include:
- Elimination of toxic organic chemicals from industrial effluents, oil spills, and solid waste.
- Sewage treatment, where microbes decompose organic matter and help remove nitrogen and phosphorus that would otherwise cause eutrophication.
- Detoxification of metals and radioactive materials such as mercury, arsenic, and uranium from soils and groundwater.
- Development of microbial biosensors that can detect pollutants through genetically engineered microbes carrying reporter genes.
The First Genetically Modified Microbe
A milestone in environmental biotechnology came with the development of the first genetically modified (GM) microbe designed for petroleum degradation. In 1972, Ananda M. Chakrabarty, working at General Electric, engineered strains of Pseudomonas putida capable of degrading multiple components of crude oil. His work led to the first U.S. patent awarded for a living organism in 1980 (Chakrabarty, 1981). Although these engineered microbes showed limited success in field applications, the innovation marked the beginning of synthetic biology approaches to environmental problems.
Microbial Degradation of Organophosphorus Pesticides
Another significant application of microbial biotechnology is the detoxification of organophosphorus (OP) pesticides, widely used in agriculture but also known as potent nerve agents. OP poisoning remains a major health problem in developing countries. Certain bacteria harbor genes such as opd (organophosphate/parathion degradation) and mpd (methyl parathion degradation), which encode enzymes capable of breaking down these pesticides into less toxic compounds (Singh & Walker, 2006). Harnessing these microbes offers a safer and more environmentally friendly alternative to chemical degradation methods.
Microbes in Wastewater Treatment
Wastewater treatment plants rely heavily on microbial communities to remove organic matter, nutrients, and pollutants before the water is released back into the environment. Two key microorganisms are central to these processes:
- Accumulibacter phosphatis – a bacterium that plays a major role in enhanced biological phosphorus removal (EBPR). It can take up excess phosphate from wastewater and store it as intracellular polyphosphate granules. This reduces the risk of eutrophication when treated water is discharged into rivers or lakes (Seviour & McIlroy, 2008).
- Nitrosomonas europaea – a chemolithoautotrophic bacterium responsible for the oxidation of ammonium (NH₄⁺) to nitrite (NO₂⁻) in the first stage of nitrification. This process is crucial for removing nitrogen compounds that would otherwise contribute to nutrient pollution (Prosser, 2005).
Microbes and Heavy Metal Detoxification
Certain microbes are naturally tolerant of heavy metals, while others have been genetically engineered to enhance their detoxification abilities. For example, Escherichia coli has been engineered to accumulate mercury ions (Hg²⁺) through the expression of synthetic, high-affinity metal-binding proteins.
- EC20 (Glu-Cys): a designed peptide with strong mercury-binding properties.
- Lpp-OmpA-EC20 fusion: anchors EC20 to the outer membrane of E. coli.
- MalE-EC20 fusion: localizes the peptide into the periplasmic space.
- MerT and MerP systems: native mercury transport proteins that enhance uptake.
These modifications enable engineered E. coli strains to bioaccumulate toxic mercury from contaminated environments, reducing its mobility and bioavailability (Roh et al., 2006).
Microbial Biosensors
Microbial biosensors are living, engineered systems designed to detect pollutants in real time. They typically involve transcriptional fusions of pollutant-responsive promoters with reporter genes such as lux (bioluminescence), gfp (green fluorescent protein), or luc (luciferase).
- Constitutive biosensors track overall cell growth or density.
- Pollutant-responsive biosensors detect specific chemicals.
One notable example is Pseudomonas fluorescens HK44, the first genetically engineered microbial biosensor released into the environment. It uses polycyclic aromatic hydrocarbon (PAH)-inducible promoters to control lux gene expression, emitting light that can be detected via fiber optic sensors (Ripp et al., 2000). Such tools are valuable for monitoring contamination in soils and groundwater.
13.1: Metagenomics – understanding microbial communities
Microorganisms (bacteria, fungi, archaea, and protozoa) are the silent wheel that functions as the ecosystem’s cradle. These microbes have been associated in one way or another with a wide array of living organisms in terrestrial and aquatic environments. They have gained the competence to function dynamically within the system. These microbes’ composition, structure, and richness are variable across different environmental systems and associated with organisms. In the case of plants, these microbes have either established endophytic (inside) or epiphytic (outside) modes of life. Prokaryotes are the most physiologically diverse and metabolically versatile organisms on our planet. Bacteria vary in how they forage for food, transduce energy, contend with competitors, and associate with allies. But the variations we know are only the tip of the microbial iceberg.  Most microorganisms have yet to be cultivated in the laboratory, and almost all our knowledge of microbial life is based on organisms raised in pure culture. The variety of the rest of the uncultured microbial world is staggering and will expand our view of what is possible in biology. The challenge that has frustrated microbiologists for decades is accessing the microorganisms that cannot be cultured in the laboratory. Many clever cultivation methods have been devised to expand the range of organisms that can be cultured. Still, knowledge of the uncultured world is slim, so it is challenging to use a process based on rational design to coax many of these organisms into the culture.
Most microorganisms have yet to be cultivated in the laboratory, and almost all our knowledge of microbial life is based on organisms raised in pure culture. The variety of the rest of the uncultured microbial world is staggering and will expand our view of what is possible in biology. The challenge that has frustrated microbiologists for decades is accessing the microorganisms that cannot be cultured in the laboratory. Many clever cultivation methods have been devised to expand the range of organisms that can be cultured. Still, knowledge of the uncultured world is slim, so it is challenging to use a process based on rational design to coax many of these organisms into the culture.
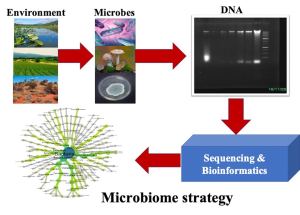
Figure 13.1. Depiction of DNA extraction from different environments to perform microbial community analysis. This helps to understand the microbial composition (bacteria and fungi) and diversity in each sample. The samples can range from soil, water, human gut, and even space environment.
13.1.1 Metagenomics
Metagenomics provides an additional set of tools to study uncultured species. This new field offers an approach to studying microbial communities as units without cultivating individual members. Metagenomics entails the extraction of DNA from an environment so that all the genomes of organisms in the microbes are pooled. These genomes are usually fragmented and cloned into an organism that can be cultured to create ‘metagenomic libraries. These libraries are then analyzed based on DNA sequences or functions conferred on the surrogate host by the metagenomic DNA. Although this field of microbiology is relatively young, discoveries have already been made that challenge existing paradigms. They have made substantial contributions to biologists’ quest to piece together the puzzle of life.
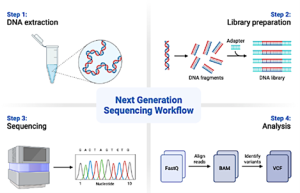
Figure 13.2. Next Generation Sequencing workflow for metagenomics (Image drawn with Biorender.com)
13.1.2 Microbiome:
A microbiome refers to the total genetic material of microbial communities associated with plants in either of the modes, whereas ‘holobiome’ or ‘holobionts’ is a sum of host and associated biota genomic material that can also include prokaryotic and eukaryotic organisms. Microorganisms (bacteria, fungi, and archaea) are the silent wheel that functions as the plant ecosystem’s cradle. These microbes’ composition, structure, and richness are variable across different environmental systems and associated with organisms. A newer concept of ‘eco-holobionts’ argues exponentially on the ecological or ecosystem-based interaction of microbiome to identify soil-plant-animal-environmental functionalities. Recently, the plant and the soil-associated microbiome has been coined as a “second genome” highly variable in diversity, abundance, and composition. Microbiome diversity and abundance in food crops alternatively influence the human gut microbiome. Healthy soil and crop microbiome are essential to healthy human growth.
13.2: Metagenomics – Methods
From a historical point of view, the term “OMICS” comes from the Greek word “Ome,” which corresponds to “whole” / “or” / “complete.” Derivatization of the concept in the form of genomes, transcriptomes, proteomes, and metabolomes, where this concept relates to cellular molecules such as genes, transcriptomes, proteins, and metabolomes. Technical interventions to study these aspects are called genomics, transcriptomics, proteomics, and metabolomics (Nalbantoglu and Karadag, 2019). The OMICS-based concept is described at various levels of the organization, beginning with “genomics” and progressing to proteomics and metabolomics. Genotyping (a sequence-based method), transcriptomics (an expression-based approach), and epigenomics (which revolves around the epigenetic regulation of genome expression) are the three subfields of genomics (Vlaanderen et al., 2010). Not only have OMICS-based approaches transformed traditional approaches to identifying microbial communities harboring extreme environmental niches, but they’ve also created next-generation platforms for understanding differential responses at the micro- to macro-level (Pan and Barrangou, 2020).
The information on the complete genome of microbial communities isolated from various environments, their interaction profile with different biotic and abiotic variables, and their biogeochemical profiling are inferred by metagenomic analysis (Figure 4(). Meanwhile, metatranscriptomics is used to identify taxonomically and functionally relevant microbiota by deciphering the expression profile of distinct mRNA transcripts in microbial communities and their tailored regulators. Metaproteomics investigations are like studying the expression profile of microbial communities’ proteomes on a broad scale. Proteogenomics, which combines metagenomics and metaproteomics, gave researchers a better grasp of the evolutionary relationships between diverse microbial communities.
13.2.1 Amplicon Sequencing and Analysis:
Amplicon sequencing is one of the new and affordable approaches to elucidate the individual and complete microbial community’s functions and attributes in unparalleled detail. The amplicon sequencing universal primers selectively bind with highly conserved sequences of desired microbial genomes in selected microbiomes (D’Amore et al., 2016). The main sequence (amplicon) used in microbiome analysis is the 16S rRNA gene. Similarly, universal primers target the small subunit rRNA genes in eukaryotes and archaea during internal transcribed spacers (ITS) in fungi (Kittelmann et al., 2013). To amplify the various hypervariable regions in the bacterial 16S rRNA, different primers have been designed and available to generate different-sized PCR products to fit the requirement of available sequencing platforms like Illumina, IonS5, Nanopore, Pacific bioscience, etc. (D’Amore et al., 2016). The microbiome sequences analyzed using amplicon sequencing are usually clustered into operational taxonomic units (OTUs) based on arbitrarily defined sequence similarity thresholds. Like the 16S rRNA gene sequences having greater than 97% sequence similarity, they are grouped and clustered into individual OTUs representing the phylogenetic borders of bacterial species. Bacterial reference genomes can be used as reference guides for sequence clustering, but de novo is a reliable approach to clustering to identify the unidentified species (Rideout et al., 2014). Similarly, fungal microbiota is analyzed using ITS amplicon sequencing methods similar to those used for bacterial 16S rRNA. On the other hand, the ITS amplicon sequences generate varying sequence lengths and similarities, sometimes complicating the clustering and classification of fungal species (Fricker et al., 2019).
The next-generation sequencing platforms, such as Illumina Miseq, which is the most used platform, require small fragments, i.e., 250-600 bp, and can generate high coverage (Kozich et al., 2013). They target hypervariable regions in the 16S rRNA gene because these are sufficient and have high accuracy in the classification. The universal primers target and amplify one or multiple hypervariable regions (V1-V9) of the 16S rRNA gene (Song and Xie, 2020). The use of these primers depends on the study design; for example, the V4 region is targeted by 515F/806R primer pair (Gilbert et al., 2014), and areas of V5-V7 by 799F/1193R primers. Multiple studies have targeted these regions to assess the microbial communities in plant soil (Bates et al., 2011), plant rhizosphere (Bates et al., 2011), and plant roots. Similarly, fungal communities are profiling using the ITS between the small subunit 18S and large subunit 28S rRNA gene. Universal primers like ITS1-F/ITS4-B amplify targeted sequences.
Furthermore, the other primers include the barcode and adapter. Alternatively, the amplicons are also sequenced using PacBio sequencing. This approach can sequence the 16S gene or large fragments of up to 30 kb (Lucaciu et al., 2019).
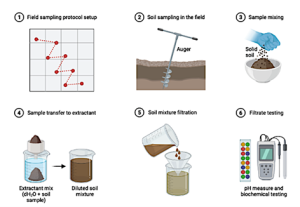
Figure 13.3. Soil sampling steps and processing for metagenomic analysis (Image drawn with Biorender.com)
Although amplicon sequencing is one of the most used approaches for microbial community profiling, it has some limitations, like sometimes various taxonomic lineage genes are amplified with unequal efficiency using universal primers (Schloss et al., 2011;Mao et al., 2012). The 16S genes that have long introns might not be amplified using normal PCR conditions because of the length size. The per genome rRNA gene clusters impact estimating a single bacterial taxa field. In addition, the databases that are compared for operational taxonomic units (OTUs) or amplicon sequence variants (ASVs) have often been marginalized for microbiomes of unique environmental systems. Many microbiome players are ‘unidentified’ or ‘unknown.’
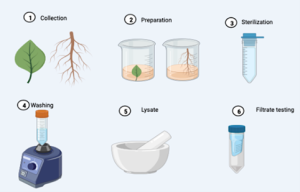
Figure 13.4. Plant samples metagenomics analysis (Image drawn with Biorender.com)
Moreover, the amplicon sequencing might affect the conclusion of community function where the functional and phylogeny are congruent. However, alternative approaches may minimize these limitations. To avoid that, several new amplicon regions have been identified in the ITS gene to ensure increased coverage and identification of variant SNPs. The larger-scale metagenome shotgun has also solved this assembly-based technology encompassing a more extensive range of bacterial and fungal OTUs or ASVs.
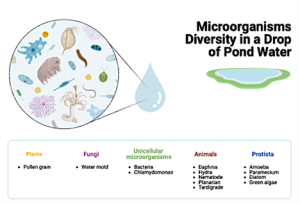
Figure 13.5. Microbial communities found in deep water (Image drawn with Biorender.com)
13.2.2 Shotgun Metagenomic Approaches:
Shotgun metagenomic is a sequencing approach used to study plant microbiomes targeting the whole or a significant proportion of microbial genomes. The primary purpose of this method is to elucidate the functional role of core microbiome players in each environmental system. The active role can be attributed to identifying several biosynthetic and metabolic pathways governing microbial function, responses, and reproduction. Using these novel mechanisms and processes can explicitly specify the functional role of microbes in extreme environmental conditions (Levy et al., 2018). For this purpose, the DNA is extracted directly from the microbial community (uncultured), and all the extracted DNA is sheared into small fragments and sequenced independently. In most cases, a 6Gb sequence data of shotgun metagenome is regarded as sufficient to elucidate the functional role of several hundreds of microbiome players. Several recent examples exist where shotgun metagenome has been sequenced for plants exposed to extreme temperature and drought conditions and elucidated with microbiome function.
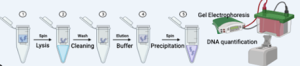
Figure 13.6. DNA extraction methods and steps for metagenomic analysis
In most cases, once the shotgun metagenome is obtained, the sequences or reads are preprocessed computationally for quality control, such as the removal of sequencing adaptors, low-quality sequences trimming, duplicate sequences removal, and foreign or non-targeted lines. This is followed by sequence analysis, performed using a combination of approaches like read-based and assemble-based, relevant to experimental design. Once the sequences are processed, different statistical techniques and tools are used to analyze and interpret data. Finally, the result of the high-dimensional biological data is validated and concluded (Quince et al., 2017). The computation sequence analysis of shotgun metagenomes is performed using four techniques: 1) taxonomic binning, 2) taxonomic profiling, 3) target–gene reassembly, and 4) genome binning.
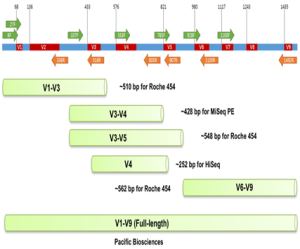
Figure 13.7. 16S rRNA gene and its analysis for different sequencing platforms
13.3: Metagenomics – Analysis
13.3.1 Taxonomic Profiling and Binning:
The resultant metagenome assemblies are highly fragmented, unknowingly comprised of thousands of contigs with relative genomes. Taxonomic profilers and binners utilize the existing databases to arrange these fragmented contigs into taxonomic groups (Quince et al., 2017). Taxonomic profilers produce per taxa abundance tables based on taxonomic groups’ presence/absence or relative abundance, like taxonomic marker-based amplicon analyses. The single copy gene marker database is used to map the read data (Truong et al., 2015), followed by matching the k-mers to the genomic databases (Hurwitz et al., 2016), or the already existing gene compositions in genomic databases are used to match gene compositions (Klingenberg and Meinicke, 2021). Similarly, taxonomic lineages are assigned to each obtained read, and the microorganisms’ relative abundance profiles in metagenomic samples are tabulated. Multiple threads can be assigned taxonomic groups using whole genome databases that can recall more taxa with low precision. Reads assigned to other Reads assigned to taxonomic groups can subsequently be mined for function or assembled independently from reads assigned to different taxonomic groups (Cleary et al., 2015). Specifying taxonomic bins at species or genus level may result in low performance otherwise can be done at any level from species to order etc. (Sczyrba et al., 2017).
13.3.2 Target Gene Assembly:
The metagenome taxonomic composition is also characterized by the reconstruction of partial/full-length rDNA sequences directly using raw data sets of the metagenomes (Miller, 2013;Gruber-Vodicka et al., 2020). Taxonomic profiling also relies on mapping the raw data to the database, but this technique differs. Target gene assembly reconstructs the full-length gene used in downstream applications like phylogenetic analysis, identification of novel taxa, and taxonomic classification (Soergel et al., 2012). This technique can also be utilized to reconstruct protein-coding function genes.
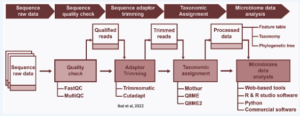
Figure 13.8. Bioinformatic workflow for metagenome data set analysis
13.3.3 Binning Genomes from Metagenomic Data Sets:
Over recent years, the recovery of metagenome-assembled genomes (MAG) from metagenomic datasets collected from complex microbial communities and microbiomes has been used widely. Initially, the genomic DNA of microbial communities is extracted and deeply sequenced. Metagenome assembly is formed to reconstruct short fragments of the underlying member genomes and further analyzed using the data clustering approach (genome binning) (Wani et al., 2022) to recover the draft genomes of the member microbial genomes. The nearly automated workflows of bioinformatics for this approach enabled the researchers to study a wide variety of microbial communities and plant microbiomes, which resulted in the recovery of numerous draft genomes of new uncharacterized microbial species, which bean remained uncharacterized because of the unknown optimal conditions required for culturing of these microbes. Despite many accomplishments, the MAGs still have challenges and limitations like limited accuracy in complex repeat regions and reconstructing sequences from closely related strains or sub-species (Cao et al., 2017).
13.3.4 Transcriptomics and Metatranscriptomics:
Transcriptomic analysis using RNA sequencing (RNA-seq) and gene expression microarray approaches are used to know the differential gene expression under different conditions of plant-associated microbes (Levy et al., 2018). RNA-seq is a very well-used method in transcriptome analysis. Metatranscriptomic was used to study the whole plant-associated microbial communities in Arabidopsis thaliana (Chaparro et al., 2014)for the first time. The RNA is extracted from plant samples, and ribosomal RNA is minimized. The most crucial step is to make cDNA from the RNA using reverse transcription. Still, the prokaryote template elongation for cDNA requires random primers, and that’s why the host plant microbiome RNA-seq the host RNA sequences are expected. The recent advancement in third-generation sequencings like PacBio and Nanopore can produce hundreds of kbp sequence read lengths, which results in the sequencing of complete transcripts. However, short-read sequencing is often used for metatranscriptomic due to low-cost Illumina.
The metatranscriptomic analysis of short read sequences can be done in assembly-based and read-based. The assembly-based transcripts can be reference-based (aligned to metagenomic bins) or reference-free (Meta transcriptomic reads only). The rRNA is removed in preprocessing RNA-seq, and low-quality bases are trimmed. The host RNA is separated in a plant microbiome by mapping the reads to one of the closely related available transcriptomes or genomes. Although there are limitations like removing rRNA, which accounts for 90% of the total RNA, mRNA is unstable.
List of tools
QIIME2 pipeline
DADA 2.0 Pipeline
Reference
Bates, S.T., Berg-Lyons, D., Caporaso, J.G., Walters, W.A., Knight, R., and Fierer, N. (2011). Examining the global distribution of dominant archaeal populations in soil. The ISME journal 5, 908-917.
Cao, Y., Fanning, S., Proos, S., Jordan, K., and Srikumar, S. (2017). A review on the applications of next generation sequencing technologies as applied to food-related microbiome studies. Frontiers in microbiology, 1829.
Chaparro, J.M., Badri, D.V., and Vivanco, J.M. (2014). Rhizosphere microbiome assemblage is affected by plant development. The ISME journal 8, 790-803.
Cleary, B., Brito, I.L., Huang, K., Gevers, D., Shea, T., Young, S., and Alm, E.J. (2015). Detection of low-abundance bacterial strains in metagenomic datasets by eigengenome partitioning. Nature biotechnology 33, 1053-1060.
D’amore, R., Ijaz, U.Z., Schirmer, M., Kenny, J.G., Gregory, R., Darby, A.C., Shakya, M., Podar, M., Quince, C., and Hall, N. (2016). A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC genomics 17, 1-20.
Fricker, A.M., Podlesny, D., and Fricke, W.F. (2019). What is new and relevant for sequencing-based microbiome research? A mini-review. Journal of advanced research 19, 105-112.
Gilbert, J., Jansson, J., and Knight, R. (2014). The Earth Microbiome project: successes and. Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R: QIIME.
Gruber-Vodicka, H.R., Seah, B.K., and Pruesse, E. (2020). phyloFlash: rapid small-subunit rRNA profiling and targeted assembly from metagenomes. Msystems 5, e00920-00920.
Hurwitz, B.L., U’ren, J.M., and Youens-Clark, K. (2016). Computational prospecting the great viral unknown. FEMS microbiology letters 363, fnw077.
Kittelmann, S., Seedorf, H., Walters, W.A., Clemente, J.C., Knight, R., Gordon, J.I., and Janssen, P.H. (2013). Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PloS one 8, e47879.
Klingenberg, H., and Meinicke, P. (2021). BinChecker: a new algorithm for quality assessment of microbial draft genomes. bioRxiv.
Kozich, J.J., Westcott, S.L., Baxter, N.T., Highlander, S.K., and Schloss, P.D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and environmental microbiology 79, 5112-5120.
Levy, A., Conway, J.M., Dangl, J.L., and Woyke, T. (2018). Elucidating Bacterial Gene Functions in the Plant Microbiome. Cell Host & Microbe 24, 475-485.
Lucaciu, R., Pelikan, C., Gerner, S.M., Zioutis, C., Köstlbacher, S., Marx, H., Herbold, C.W., Schmidt, H., and Rattei, T. (2019). A bioinformatics guide to plant microbiome analysis. Frontiers in plant science 10, 1313.
Mao, D.-P., Zhou, Q., Chen, C.-Y., and Quan, Z.-X. (2012). Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC microbiology 12, 1-8.
Miller, C.S. (2013). “Assembling full-length rRNA genes from short-read metagenomic sequence datasets using EMIRGE,” in Methods in Enzymology. Elsevier), 333-352.
Nalbantoglu, S., and Karadag, A. (2019). Introductory chapter: Insight into the OMICS technologies and molecular medicine. Molecular Medicine.
Pan, M., and Barrangou, R. (2020). Combining omics technologies with CRISPR-based genome editing to study food microbes. Current opinion in biotechnology 61, 198-208.
Quince, C., Walker, A.W., Simpson, J.T., Loman, N.J., and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nature Biotechnology 35, 833-844.
Rideout, J.R., He, Y., Navas-Molina, J.A., Walters, W.A., Ursell, L.K., Gibbons, S.M., Chase, J., Mcdonald, D., Gonzalez, A., and Robbins-Pianka, A. (2014). Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2, e545.
Schloss, P.D., Gevers, D., and Westcott, S.L. (2011). Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PloS one 6, e27310.
Sczyrba, A., Hofmann, P., Belmann, P., Koslicki, D., Janssen, S., Dröge, J., Gregor, I., Majda, S., Fiedler, J., and Dahms, E. (2017). Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nature methods 14, 1063-1071.
Soergel, D.A., Dey, N., Knight, R., and Brenner, S.E. (2012). Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. The ISME journal 6, 1440-1444.
Song, L., and Xie, K. (2020). Engineering CRISPR/Cas9 to mitigate abundant host contamination for 16S rRNA gene-based amplicon sequencing. Microbiome 8, 1-15.
Truong, D.T., Franzosa, E.A., Tickle, T.L., Scholz, M., Weingart, G., Pasolli, E., Tett, A., Huttenhower, C., and Segata, N. (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nature methods 12, 902-903.
Vlaanderen, J., Moore, L.E., Smith, M.T., Lan, Q., Zhang, L., Skibola, C.F., Rothman, N., and Vermeulen, R. (2010). Application of OMICS technologies in occupational and environmental health research; current status and projections. Occupational and environmental medicine 67, 136-143.
Wani, A.K., Akhtar, N., Singh, R., Chopra, C., Kakade, P., Borde, M., Al-Khayri, J.M., Suprasanna, P., and Zimare, S.B. (2022). Prospects of advanced metagenomics and meta-omics in the investigation of phytomicrobiome to forecast beneficial and pathogenic response. Molecular Biology Reports, 1-15.
Jaiswal, S., & Shukla, P. (2020). Alternative strategies for microbial remediation of pollutants via synthetic biology. Frontiers in Microbiology, 11, 808. https://doi.org/10.3389/fmicb.2020.00808
Singh, B. K., & Walker, A. (2006). Microbial degradation of organophosphorus compounds. FEMS Microbiology Reviews, 30(3), 428–471.
Bryant, R. S., & Burchfield, T. E. (1989). Review of microbial enhanced oil recovery (MEOR) field tests and projects. SPE Reservoir Engineering, 4(01), 151–154.
Demirbas, A., & Demirbas, M. F. (2011). Importance of algae oil as a source of biodiesel. Energy Conversion and Management, 52(1), 163–170.
Ripp, S., Nivens, D. E., Ahn, Y., Werner, C., Jarrell, J., Easter, J. P., … & Sayler, G. S. (2000). Controlled field release of a bioluminescent genetically engineered microorganism for bioremediation process monitoring and control. Environmental Science & Technology, 34(5), 846–853.
Seviour, R. J., & McIlroy, S. J. (2008). The microbiology of biological phosphorus removal in activated sludge systems. FEMS Microbiology Reviews, 32(3), 585–617.
